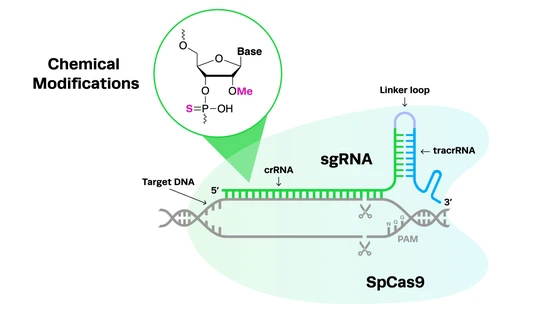
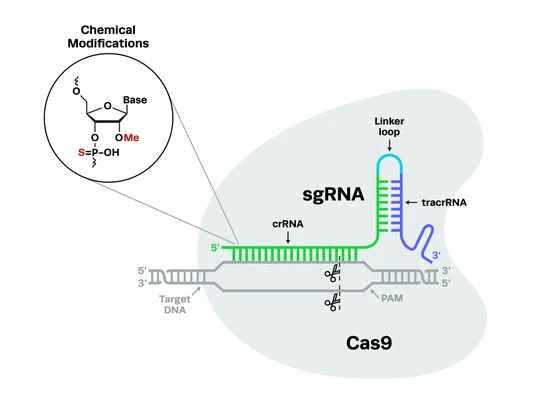
The field of cell and gene therapies is constantly evolving, and there has been significant recent progress in this exciting area of research. Yet despite the promise of these therapies, the regulations governing them seem to lag behind, hindering the clinical translation of these novel medicines. There have been several recent indications that the FDA plans to provide greater support for cell and gene therapies, addressing key issues that continue to impede their development.
The recent American Society of Gene and Cell Therapy (ASGCT) meeting hosted some interesting discussions on current regulatory trends, providing some critical insight into problems in the cell and gene therapy field, expected growth, new FDA initiatives, and attempts to bridge gaps between regulators and researchers. In this blog, we’ll provide a recap of some of the critical points of discussion concerning the cell and gene therapy (CGT) regulatory landscape from the ASGCT meeting.
Recent Problems in Cell & Gene Therapy Development
There has been an obvious lull in CGT product approvals in the last two years, and this downturn has been driven by several key factors. Where there were already several impediments to getting cell and gene therapies approved by the FDA, these issues have been exacerbated by the global COVID-19 pandemic.
Bottlenecks in the manufacturing process, communication issues between the FDA and sponsors, and shortages of qualified specialist staff have caused many therapies to falter at critical stages of the clinical development pipeline. In addition to the challenges faced by researchers and companies, the FDA is known to be chronically understaffed, making it difficult to get therapies through the review process in a timely manner.
Another key problem in the industry is the financial feasibility of generating small-batch CGT products for ‘ultra-rare’ diseases with limited patient populations. Many companies are unable to develop these niche therapies because of the associated costs and limited financial return.
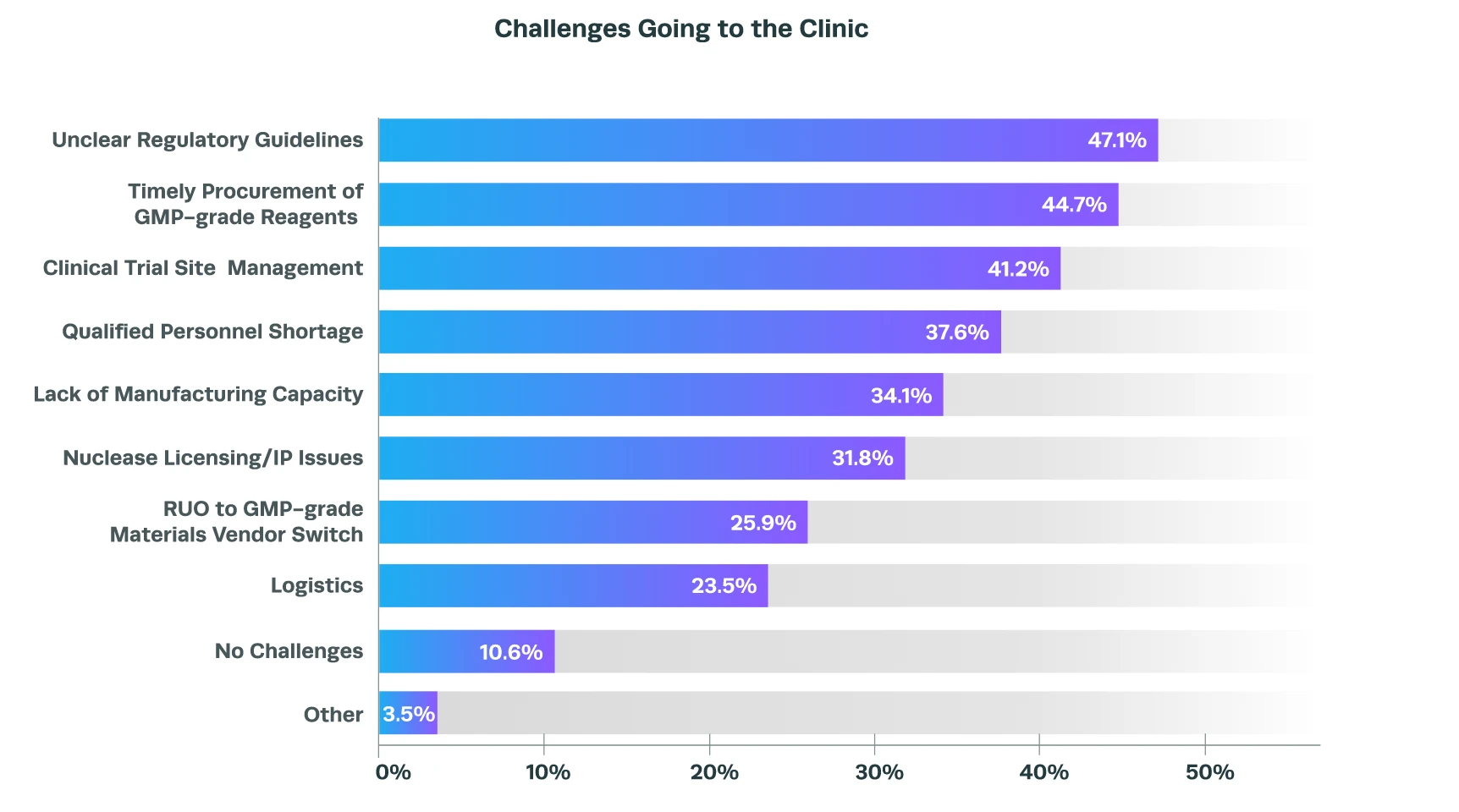
Our recent survey revealed that dealing with confusing regulatory guidelines is one of the top obstacles faced by researchers developing CGT products, closely followed by the accessibility of good manufacturing practice (GMP)-grade reagents and issues with clinical trial site management. For more information about these and other issues, you can read our blog on the top 5 hurdles faced by CRISPR cell and gene therapy researchers when progressing into the clinic.
CRISPR Cell and Gene Therapies: Key Challenges in Clinical Development
Researchers face many hurdles when developing CRISPR therapy products, including the procurement of true GMP reagents, unclear regulatory guidelines, lack of consistency during development, and shortages of qualified experts. Read more about these issues in CRISPR therapy development and strategies to overcome them.

Current Regulatory Trends in Cell & Gene Therapy
The current regulatory landscape of cell and gene therapies is undergoing significant changes, many of which were discussed at the 2022 ASGCT meeting. In this section, we’ll briefly discuss some major recent developments, including several FDA-led initiatives and new draft guidance for specific types of therapies.
Expected growth in cell and gene therapy IND filingsAccording to Wilson Bryan, the director of the FDA’s Office of Tissues and Advanced Therapies (OTAT), the number of CGT investigational new drug (IND) filings submitted to the FDA dropped from 350 in 2020 down to 299 in 2021. However, he remarked that these numbers are expected to increase again in the coming years.
The expected growth in 2022 and beyond is in large part due to the FDA’s Cellular, Tissue, and Gene Therapies Advisory Committee (CTGTAC) panel recently voting to approve two gene-edited cell therapies from biotech company Bluebird Bio. While the final FDA approval is still pending, the unanimous votes in favor of these therapies are strong indicators that they will be successful in the final stage. Despite several early-stage setbacks, the FDA’s recent support for Bluebird’s viral vector gene therapy products lays the regulatory groundwork for developers of similar CGT therapies and indicates a higher likelihood of their success.
Also noted by Bryan is the recent increase in the number of Breakthrough (BT) and Regenerative Medicine Advanced Therapy (RMAT) designation requests, applications for which are made alongside IND submission or during an existing IND filing. There has also been an increase in the number of RMAT requests that have been accepted. BT and RMAT designations provide similar benefits to fast track designation, however, they also require higher levels of evidence to support their status in either of these categories.
According to Wilson Bryan, the director of the FDA’s Office of Tissues and Advanced Therapies (OTAT), the number of CGT investigational new drug (IND) filings submitted to the FDA dropped from 350 in 2020 down to 299 in 2021. However, he remarked that these numbers are expected to increase again in the coming years.
The expected growth in 2022 and beyond is in large part due to the FDA’s Cellular, Tissue, and Gene Therapies Advisory Committee (CTGTAC) panel recently voting to approve two gene-edited cell therapies from biotech company Bluebird Bio. While the final FDA approval is still pending, the unanimous votes in favor of these therapies are strong indicators that they will be successful in the final stage. Despite several early-stage setbacks, the FDA’s recent support for Bluebird’s viral vector gene therapy products lays the regulatory groundwork for developers of similar CGT therapies and indicates a higher likelihood of their success.
Also noted by Bryan is the recent increase in the number of Breakthrough (BT) and Regenerative Medicine Advanced Therapy (RMAT) designation requests, applications for which are made alongside IND submission or during an existing IND filing. There has also been an increase in the number of RMAT requests that have been accepted. BT and RMAT designations provide similar benefits to fast track designation, however, they also require higher levels of evidence to support their status in either of these categories.
The Bespoke Gene Therapy ConsortiumThe FDA recently announced a new initiative as part of its NIH Accelerating Medicines Partnership Program. The Bespoke Gene Therapy Consortium (BGTC) aims to overcome major obstacles and streamline the development process for small-batch gene therapies.
As described in the ASGCT meeting by Peter Marks, director of the FDA’s Center for Biologics Evaluation and Research (CBER), the BGTC will act as a ‘cookbook’ for these therapies, providing information on basic and clinical research, manufacturing, and production, and regulatory requirements.
While each ultra-rare genetic disorder may have a very small patient population, when these conditions are grouped together, they make up a significant number. The FDA has indicated that financial incentives are also required in order to encourage companies to create therapies that are not ‘commercially viable’.
The idea of generating gene therapies for very small patient populations has been championed by several researchers in the CRISPR field, including Professor Fyodor Urnov of UC Berkeley’s Innovative Genomics Institute. Urnov has also suggested that an entirely new clinical framework is required for edited CGTs in order to get treatments to patients faster.
For more of Professor Urnov’s perspective on CRISPR cell and gene therapies for ultra-rare diseases, you can watch his keynote address at World CRISPR Day 2021.
The FDA recently announced a new initiative as part of its NIH Accelerating Medicines Partnership Program. The Bespoke Gene Therapy Consortium (BGTC) aims to overcome major obstacles and streamline the development process for small-batch gene therapies.
As described in the ASGCT meeting by Peter Marks, director of the FDA’s Center for Biologics Evaluation and Research (CBER), the BGTC will act as a ‘cookbook’ for these therapies, providing information on basic and clinical research, manufacturing, and production, and regulatory requirements.
While each ultra-rare genetic disorder may have a very small patient population, when these conditions are grouped together, they make up a significant number. The FDA has indicated that financial incentives are also required in order to encourage companies to create therapies that are not ‘commercially viable’.
The idea of generating gene therapies for very small patient populations has been championed by several researchers in the CRISPR field, including Professor Fyodor Urnov of UC Berkeley’s Innovative Genomics Institute. Urnov has also suggested that an entirely new clinical framework is required for edited CGTs in order to get treatments to patients faster.
For more of Professor Urnov’s perspective on CRISPR cell and gene therapies for ultra-rare diseases, you can watch his keynote address at World CRISPR Day 2021.
INTERACT meetings and a gene therapy pilot programA relatively recent initiative by the FDA is the INitial Targeted Engagement for Regulatory Advice on CBER producTs (INTERACT) program. This program is essentially an attempt to address the growing pains experienced by the CGT industry within current clinical frameworks and involves informal meetings between CBER staff and researchers/sponsors who are at the pre-IND stage of development.
During his ASGCT address, Marks also commented on another new initiative called the Gene Therapy Pilot Program. According to Marks, this program would involve the FDA providing real-time feedback to sponsors throughout the clinical development process. It remains to be seen how plausible this level of extensive feedback will be, considering the FDA’s current staffing issues.
A relatively recent initiative by the FDA is the INitial Targeted Engagement for Regulatory Advice on CBER producTs (INTERACT) program. This program is essentially an attempt to address the growing pains experienced by the CGT industry within current clinical frameworks and involves informal meetings between CBER staff and researchers/sponsors who are at the pre-IND stage of development.
During his ASGCT address, Marks also commented on another new initiative called the Gene Therapy Pilot Program. According to Marks, this program would involve the FDA providing real-time feedback to sponsors throughout the clinical development process. It remains to be seen how plausible this level of extensive feedback will be, considering the FDA’s current staffing issues.
New draft guidance for gene therapies and CAR-T cell therapiesIn March of 2022, the FDA released two new draft guidance documents to help sponsors who are developing CGT products. The first is titled ‘Human Gene Therapy Products Incorporating Human Genome Editing’, and provides recommendations on the information that should be included in IND filings for these therapies, including study design, safety, and manufacturing.
The second draft guidance is ‘Considerations for the Development of Chimeric Antigen Receptor (CAR) T Cell Products’. While the title refers specifically to CAR-T cell therapies, which are used to treat cancer, the FDA remarked that these guidelines are also more broadly applicable to other gene-edited cell therapies, such as natural killer (NK) and T cell receptor (TCR) cell therapies. The document includes recommendations on safety, manufacturing, clinical study design, and analytical comparability of CAR-T products.
In March of 2022, the FDA released two new draft guidance documents to help sponsors who are developing CGT products. The first is titled ‘Human Gene Therapy Products Incorporating Human Genome Editing’, and provides recommendations on the information that should be included in IND filings for these therapies, including study design, safety, and manufacturing.
The second draft guidance is ‘Considerations for the Development of Chimeric Antigen Receptor (CAR) T Cell Products’. While the title refers specifically to CAR-T cell therapies, which are used to treat cancer, the FDA remarked that these guidelines are also more broadly applicable to other gene-edited cell therapies, such as natural killer (NK) and T cell receptor (TCR) cell therapies. The document includes recommendations on safety, manufacturing, clinical study design, and analytical comparability of CAR-T products.
Regulatory harmonization and agreements on surrogate endpointsA major topic of discussion during the ASGCT panel discussion was the concept of regulatory harmonization. As Ana Hidalgo-Simon of the European Medicines Agency (EMA) commented, there are significant problems in multi-country clinical trials due to the different requirements of the parties involved. A complicating factor is that the definitions of CGT products and their components differ between countries, making it difficult to achieve any consensus on their regulation. Harmonization of CGT requirements and improved inter-country communication are key for the progression of the field throughout Europe.
Lack of agreement on surrogate endpoints in clinical trials was another key theme during the session. The FDA’s accelerated approval program, which allows for earlier approval of new therapies that address unmet medical needs, is based on surrogate endpoints. These endpoints are markers that can predict clinical benefit but are not actual measurements of clinical benefit as would be expected from standard trials.
During the discussions, Marks noted that accelerated approval using surrogate endpoints is necessary to develop gene therapy products for rare disease populations, however, the FDA often does not agree to surrogate endpoints being accurate predictors of the clinical benefit of a therapy before the trial begins. This lack of agreement between the FDA and trial sponsors leads to significant problems at later stages of clinical trials. Marks indicated that this issue needs to be addressed in order for CGT products to have reasonable chances of success.
A major topic of discussion during the ASGCT panel discussion was the concept of regulatory harmonization. As Ana Hidalgo-Simon of the European Medicines Agency (EMA) commented, there are significant problems in multi-country clinical trials due to the different requirements of the parties involved. A complicating factor is that the definitions of CGT products and their components differ between countries, making it difficult to achieve any consensus on their regulation. Harmonization of CGT requirements and improved inter-country communication are key for the progression of the field throughout Europe.
Lack of agreement on surrogate endpoints in clinical trials was another key theme during the session. The FDA’s accelerated approval program, which allows for earlier approval of new therapies that address unmet medical needs, is based on surrogate endpoints. These endpoints are markers that can predict clinical benefit but are not actual measurements of clinical benefit as would be expected from standard trials.
During the discussions, Marks noted that accelerated approval using surrogate endpoints is necessary to develop gene therapy products for rare disease populations, however, the FDA often does not agree to surrogate endpoints being accurate predictors of the clinical benefit of a therapy before the trial begins. This lack of agreement between the FDA and trial sponsors leads to significant problems at later stages of clinical trials. Marks indicated that this issue needs to be addressed in order for CGT products to have reasonable chances of success.
Improving communication between FDA and sponsorsCommunication issues between sponsors and the FDA is an ongoing issue in the development of cell and gene therapies. For many researchers and sponsors, progressing from pre-clinical research into clinical trials can be an extremely challenging process, not least because of unclear regulatory guidelines. Fortunately, it seems the FDA is now making it a priority to more effectively communicate with those developing CGT products, encouraging a growth trajectory in the coming years.
OTAT recently announced that it plans to provide increased guidance for sponsors, facilitating a variety of workshops and webinars. Increased consistency in the review process and timeliness of reviews have also been touted as key priorities, along with broader public messaging surrounding these therapies. Through these initiatives and further engagement with the industry, the FDA hopes to enable the development and, most importantly, success, of more complex CGT products, bringing them to patients sooner.
The next several years are likely to hold some interesting developments in the world of cell and gene therapy. The FDA’s recent surge of support for these therapies, along with an increase in the number of CGT products progressing towards clinical trials, will hopefully result in an increased number of therapies making it through the final stages of clinical trials and gaining FDA approval.
Communication issues between sponsors and the FDA is an ongoing issue in the development of cell and gene therapies. For many researchers and sponsors, progressing from pre-clinical research into clinical trials can be an extremely challenging process, not least because of unclear regulatory guidelines. Fortunately, it seems the FDA is now making it a priority to more effectively communicate with those developing CGT products, encouraging a growth trajectory in the coming years.
OTAT recently announced that it plans to provide increased guidance for sponsors, facilitating a variety of workshops and webinars. Increased consistency in the review process and timeliness of reviews have also been touted as key priorities, along with broader public messaging surrounding these therapies. Through these initiatives and further engagement with the industry, the FDA hopes to enable the development and, most importantly, success, of more complex CGT products, bringing them to patients sooner.
The next several years are likely to hold some interesting developments in the world of cell and gene therapy. The FDA’s recent surge of support for these therapies, along with an increase in the number of CGT products progressing towards clinical trials, will hopefully result in an increased number of therapies making it through the final stages of clinical trials and gaining FDA approval.