CRISPR cell and gene therapies hold immense promise but navigating their unique regulatory challenges can be complex. Synthego simplifies this process, offering expert guidance in every phase - from CMC Module 3 preparation to direct interactions with regulators like the FDA and EMA. Our team stays ahead of evolving CRISPR regulations, ensuring your GMP CRISPR molecules and regulatory submissions meet the highest standards. By partnering with Synthego, you can smoothly transition your therapy from the lab to the clinic with the support and expertise needed to advance groundbreaking science.
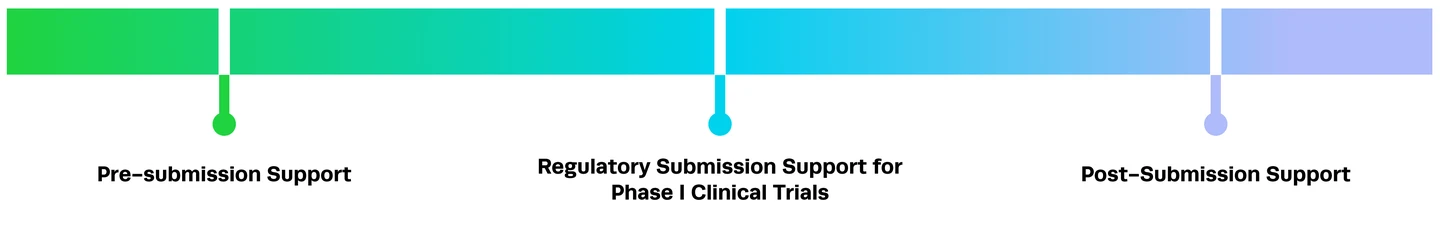

CRISPR in the Clinic: Synthego’s Regulatory Experts Discuss Development of Cell and Gene Therapies
The CRISPR gene therapy regulatory space is changing rapidly. In this blog, we’re shining a light on regulatory scientists, the unsung heroes of clinical development.