Contents
"However bad life may seem, there is always something you can do, and succeed at. While there’s life, there is hope."
Stephen Hawking
Professor Stephen Hawking amazed the world not only because of his intellect but the fact that he lived and worked for five decades with a disease that was predicted to kill him within two years: amyotrophic lateral sclerosis, or ALS.
Many people will be aware of amyotrophic lateral sclerosis because of the work of Professor Hawking or the music of famed guitar virtuoso and ALS patient Jason Becker. The condition also garnered international attention in the last decade owing to the viral ice bucket challenge that swept social media in an effort to raise funds for ALS research. But few outside the ALS community are familiar with the details and impact of this devastating neurodegenerative disease.
Significant advances in CRISPR gene editing technology and a slew of resulting studies have recently renewed hope for ALS patients and their families. In this blog, we’re discussing ALS symptoms, diagnosis, and treatment options, the current state of ALS research and clinical trials, and exploring the impact of CRISPR technology in ALS research, including improved disease modeling and the potential for curative gene therapies.
What is Amyotrophic Lateral Sclerosis?
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that causes the degeneration and eventual death of nerve cells known as motor neurons. Because motor neurons normally send signals from the brain to the muscles to control movement, patients with ALS experience severe muscle wasting. As a result, they suffer a gradual loss of coordination and movement, eventually losing the ability to walk, speak, eat, and breathe. This devastating disease has no cure and is always fatal; patients typically die within two to five years of being diagnosed.
In the Greek language, amyotrophic can be translated to ‘no muscle nourishment’, referring to the wasting of muscle from the disease, while lateral refers to the region of the spinal cord in which the affected nerve cells reside. Sclerosis is the process of scarring and hardening that occurs in the affected areas of the nervous system. ALS is often also referred to as motor neuron disease (MND) or Lou Gehrig’s disease, after the famous Yankees baseball player who had the condition. According to the ALS Association, around 5,000 people are diagnosed with ALS each year, and someone dies from the condition every 90 minutes.
NIH Scientists Discuss the iNDI Project for Alzheimer’s Disease Modeling
The Inducible Pluripotent Stem Cell Neurodegeneration Initiative (iNDI), an NIH effort, aims to standardize disease models for Alzheimer’s and related dementias. Hear the principal contributors, Dr. Michael E. Ward and Dr. Mark R. Cookson, scientists at the NIH, talk about the inception of the iNDI project, the challenges and opportunities, and the future of neurodegenerative research.

Amyotrophic Lateral Sclerosis: Symptoms, Diagnosis, and Treatment
Before we explore the current state of amyotrophic lateral sclerosis research and the role of CRISPR technology in this disease area, let’s discuss the basics of ALS, including the symptoms and progression, diagnosis, and current treatment options for this disease.
Amyotrophic lateral sclerosis symptoms and progressionThere are a range of symptoms of ALS. Typical early signs of the disease include weakness in hands, feet, or legs, often accompanied by involuntary muscle twitches and spasms. Slurred speech, trouble swallowing, and other movement difficulties are also common symptoms.
The progressive nature of ALS means that these symptoms become worse over time as the muscles atrophy. In the later stages of the disease, all basic motor functions are impaired, including breathing, speaking, and eating. Progression of the disease is also frequently accompanied by cognitive and behavioral changes.
Compared to other neurodegenerative diseases, the progression of ALS is rapid. A majority of patients die within two years of the first onset of symptoms, however, some people with ALS can live for 10 years or longer. The lifespan of patients is now known to be correlated with the age of onset - people who are younger at the time of onset tend to live longer. Stephen Hawking, for example, was 21 when he discovered he had ALS, and he lived for more than 50 years after his diagnosis.
There are a range of symptoms of ALS. Typical early signs of the disease include weakness in hands, feet, or legs, often accompanied by involuntary muscle twitches and spasms. Slurred speech, trouble swallowing, and other movement difficulties are also common symptoms.
The progressive nature of ALS means that these symptoms become worse over time as the muscles atrophy. In the later stages of the disease, all basic motor functions are impaired, including breathing, speaking, and eating. Progression of the disease is also frequently accompanied by cognitive and behavioral changes.
Compared to other neurodegenerative diseases, the progression of ALS is rapid. A majority of patients die within two years of the first onset of symptoms, however, some people with ALS can live for 10 years or longer. The lifespan of patients is now known to be correlated with the age of onset - people who are younger at the time of onset tend to live longer. Stephen Hawking, for example, was 21 when he discovered he had ALS, and he lived for more than 50 years after his diagnosis.
Diagnosis of amyotrophic lateral sclerosisUnfortunately, there is no single test, such as a biomarker-based blood test, that can accurately diagnose ALS. Doctors may first perform blood tests or take a urine sample or muscle biopsy to rule out other possible causes of symptoms. Other tests may include physical exams, MRI of the brain and spinal cord, electromyography (EMG) to measure the electrical activity of the muscle fibers, and nerve conduction studies (NCS).
Unfortunately, there is no single test, such as a biomarker-based blood test, that can accurately diagnose ALS. Doctors may first perform blood tests or take a urine sample or muscle biopsy to rule out other possible causes of symptoms. Other tests may include physical exams, MRI of the brain and spinal cord, electromyography (EMG) to measure the electrical activity of the muscle fibers, and nerve conduction studies (NCS).
Amyotrophic lateral sclerosis treatment optionsCurrent treatment options for ALS are extremely limited, and no therapies are able to repair damaged neurons or prevent disease progression. The two drugs that are available can slow the decline of ALS patients or prolong survival by short timeframes. Other treatments involve physical therapy and rehabilitation, and support for communication and breathing.
Current treatment options for ALS are extremely limited, and no therapies are able to repair damaged neurons or prevent disease progression. The two drugs that are available can slow the decline of ALS patients or prolong survival by short timeframes. Other treatments involve physical therapy and rehabilitation, and support for communication and breathing.
What Causes Amyotrophic Lateral Sclerosis?

Amyotrophic lateral sclerosis is an incredibly complex disease and in many ways is not well understood, making it all the more difficult to treat. While 90% of ALS cases are sporadic and have unknown causes, the remaining 10% are caused by genetic mutations and are hereditary, known as familial ALS (fALS). Let’s explore the genetic and environmental factors that contribute to ALS.
Genetic mutationsWhile there are more than 50 genes implicated in ALS, mutations in only a few genes have been confirmed to cause the disease, including chromosome 9 open reading frame 72 (C9orf72), cytosolic Cu/Zn superoxide dismutase (SOD1), TAR DNA binding protein 43 (TARDBP), and fused in sarcoma (FUS). More recently, another gene, serine palmitoyltransferase long chain base subunit 1 (SPTLC1), has also been confirmed to cause a rare childhood form of ALS. More genetic causes for fALS are being discovered as scientists continue to study the disease.
While there are more than 50 genes implicated in ALS, mutations in only a few genes have been confirmed to cause the disease, including chromosome 9 open reading frame 72 (C9orf72), cytosolic Cu/Zn superoxide dismutase (SOD1), TAR DNA binding protein 43 (TARDBP), and fused in sarcoma (FUS). More recently, another gene, serine palmitoyltransferase long chain base subunit 1 (SPTLC1), has also been confirmed to cause a rare childhood form of ALS. More genetic causes for fALS are being discovered as scientists continue to study the disease.
C9orf72Mutations in C9orf72 are responsible for between 35% and 45% of fALS cases. The mutation is a hexanucleotide GGGGCC repeat hyperexpansion, located in the first intron of the gene. In healthy individuals, there are typically 20 or fewer GGGGAA repeats, however, in fALS patients, the hyperexpansion can contain hundreds of repeats. This hyperexpansion disrupts the promoter region that would normally control the transcription of the C9orf72 gene. While it is unclear exactly how the mutation causes disease onset, overexpression and accumulation of the protein produced by C9orf72 can contribute to cellular dysfunction.
Mutations in C9orf72 are responsible for between 35% and 45% of fALS cases. The mutation is a hexanucleotide GGGGCC repeat hyperexpansion, located in the first intron of the gene. In healthy individuals, there are typically 20 or fewer GGGGAA repeats, however, in fALS patients, the hyperexpansion can contain hundreds of repeats. This hyperexpansion disrupts the promoter region that would normally control the transcription of the C9orf72 gene. While it is unclear exactly how the mutation causes disease onset, overexpression and accumulation of the protein produced by C9orf72 can contribute to cellular dysfunction.
SOD1The SOD1 gene encodes a protein called Cu, Zn, superoxide dismutase, which is a neuroprotective enzyme. As many as 170 different mutations in the SOD1 gene can cause fALS, disrupting cellular homeostasis through a variety of mechanisms. These mechanisms are currently not well understood, as even patients carrying the same SOD1 mutation can have differences in disease phenotype.
The SOD1 gene encodes a protein called Cu, Zn, superoxide dismutase, which is a neuroprotective enzyme. As many as 170 different mutations in the SOD1 gene can cause fALS, disrupting cellular homeostasis through a variety of mechanisms. These mechanisms are currently not well understood, as even patients carrying the same SOD1 mutation can have differences in disease phenotype.
TARDBPTARDBP encodes the DNA/RNA binding TDP-43 protein, which is able to move between the nucleus and the cytoplasm of cells and plays a key role in the splicing, transport, and translation of RNA. It is also known to be involved in important post-translational modifications such as phosphorylation and ubiquitination. Up to 50 different mutations in TARDBP can cause fALS; both the lack of TDP-43 and the overexpression of TDP-43 within cells can contribute to the disease.
TARDBP encodes the DNA/RNA binding TDP-43 protein, which is able to move between the nucleus and the cytoplasm of cells and plays a key role in the splicing, transport, and translation of RNA. It is also known to be involved in important post-translational modifications such as phosphorylation and ubiquitination. Up to 50 different mutations in TARDBP can cause fALS; both the lack of TDP-43 and the overexpression of TDP-43 within cells can contribute to the disease.
FUSThe function of the FUS gene and the protein it produces are not well understood, although it is known to contribute to RNA splicing and trafficking, DNA repair, and cellular stress responses. There have been more than 50 FUS mutations described that contribute to fALS, particularly juvenile fALS. These mutations cluster within specific regions of the gene, and typically cause mislocation of FUS protein within cells, contributing to degeneration of neurons.
The function of the FUS gene and the protein it produces are not well understood, although it is known to contribute to RNA splicing and trafficking, DNA repair, and cellular stress responses. There have been more than 50 FUS mutations described that contribute to fALS, particularly juvenile fALS. These mutations cluster within specific regions of the gene, and typically cause mislocation of FUS protein within cells, contributing to degeneration of neurons.
SPTLC1In 2021, scientists discovered that mutations in the SPTLC1 gene cause juvenile-onset fALS. The protein encoded by this gene is an enzyme involved in the biosynthesis of sphingolipids, which play a role in membrane biology and regulate cell function. Several SPTLC1 mutations were confirmed to cause juvenile fALS in the study and research is ongoing.
In 2021, scientists discovered that mutations in the SPTLC1 gene cause juvenile-onset fALS. The protein encoded by this gene is an enzyme involved in the biosynthesis of sphingolipids, which play a role in membrane biology and regulate cell function. Several SPTLC1 mutations were confirmed to cause juvenile fALS in the study and research is ongoing.
UBQLN2/PEG10In a major breakthrough, a 2023 paper revealed a previously unknown cause of ALS: paternally expressed gene 10 (PEG10). PEG10 is a fragment of an ancient retrovirus that integrated into the human genome millions of years ago. Since it can no longer replicate, the gene does not cause disease, but is still passed on from generation to generation. This domesticated retrotransposon has previously been shown to be involved in placental development in mammals.
Interestingly, however, when the PEG10 protein is overexpressed in the wrong areas - such as the spinal cord - it interferes with the expression of axon remodeling genes, preventing nerve cells from communicating properly. This overexpression of PEG10 protein seems to occur due to a previously identified mutation in the ubiquilin-2 gene (UBQLN2), a proteasome shuttle that normally regulates the expression of domesticated retrotransposons. It’s likely that PEG10 will now be developed as a biomarker for ALS because of its overabundance in ALS patients.
In a major breakthrough, a 2023 paper revealed a previously unknown cause of ALS: paternally expressed gene 10 (PEG10). PEG10 is a fragment of an ancient retrovirus that integrated into the human genome millions of years ago. Since it can no longer replicate, the gene does not cause disease, but is still passed on from generation to generation. This domesticated retrotransposon has previously been shown to be involved in placental development in mammals.
Interestingly, however, when the PEG10 protein is overexpressed in the wrong areas - such as the spinal cord - it interferes with the expression of axon remodeling genes, preventing nerve cells from communicating properly. This overexpression of PEG10 protein seems to occur due to a previously identified mutation in the ubiquilin-2 gene (UBQLN2), a proteasome shuttle that normally regulates the expression of domesticated retrotransposons. It’s likely that PEG10 will now be developed as a biomarker for ALS because of its overabundance in ALS patients.
Environmental factorsThe vast majority of ALS cases are sporadic, and the cause is unknown. However, there are environmental factors that are thought to place people at higher risk of developing the disease. These factors include exposure to toxins such as lead, and smoking, particularly in women. Individuals who have served in the military are also at a higher risk of developing ALS, although it is unclear exactly why this is the case.
The vast majority of ALS cases are sporadic, and the cause is unknown. However, there are environmental factors that are thought to place people at higher risk of developing the disease. These factors include exposure to toxins such as lead, and smoking, particularly in women. Individuals who have served in the military are also at a higher risk of developing ALS, although it is unclear exactly why this is the case.
Amyotrophic lateral sclerosis research and clinical trials
One of the notable results of the ALS ice bucket challenge was the approval of the new drug AMX0035. The combination drug, developed by Amylyx, was pushed through a pilot trial and later phase clinical trials with funds from the ALS Association raised by the challenge. Although controversial, it was approved by the FDA prior to the completion of phase III trials on the basis that it could extend the lifespan of ALS patients by several months.
Currently, a significant amount of ALS research and clinical trials is focused on antisense oligonucleotide (ASO) technology. ASOs target specific genes in order to change the function or reduce the expression of the gene - for example, the genes that are mutated in ALS. Tofersen, developed by Ionis, was the first ASO to receive accelerated approval from the FDA to treat ALS by targeting the mutated SOD1 gene.
Despite decades of ALS research, there remain limited treatment options and no curative therapies. However, there is now new hope for ALS patients and their families thanks to CRISPR technology.
CRISPR ALS Breakthroughs: Disease Modeling and Gene Therapies
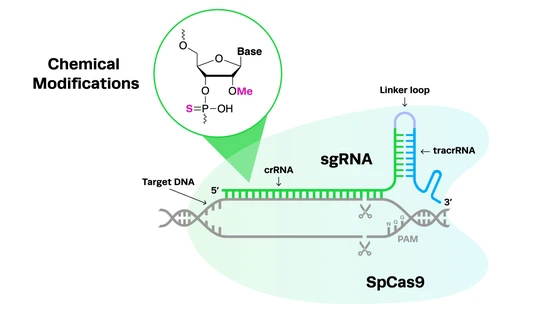
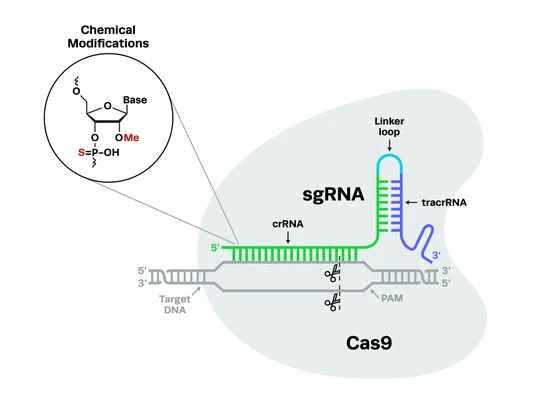
CRISPR-Cas9 technology has led to some key breakthroughs in amyotrophic lateral sclerosis research by creating more accurate disease models, revealing new drug targets, and creating potentially curative therapies. Let’s take a look at some of the key developments in CRISPR ALS disease modeling and gene therapy.
CRISPR ALS disease modelingCompared to previous genome engineering technologies, CRISPR-Cas9 can be used to edit DNA with relative ease, particularly in primary cells such as stem cells and neurons. This has been crucial in ALS research, where accurate disease models are desperately needed in order to both understand the cause and progression of the condition, identify novel drug targets, and test new treatment options before they progress into human trials.
Compared to previous genome engineering technologies, CRISPR-Cas9 can be used to edit DNA with relative ease, particularly in primary cells such as stem cells and neurons. This has been crucial in ALS research, where accurate disease models are desperately needed in order to both understand the cause and progression of the condition, identify novel drug targets, and test new treatment options before they progress into human trials.
Genome-wide CRISPR screens reveal new ALS drug targetPublished in late 2022 by researchers from Stanford University School of Medicine, a key study used CRISPR-Cas9 to perform a genome-wide screen in human cells. The paper was able to identify genes that code for lysosomal vacuolar ATPase (v-ATPase) that can lower levels of ataxin-2, a protein involved in ALS. Since several small molecule inhibitors of v-ATPase are already approved to treat other conditions, this is likely to yield positive results for ALS patients within relatively short timeframes.
Published in late 2022 by researchers from Stanford University School of Medicine, a key study used CRISPR-Cas9 to perform a genome-wide screen in human cells. The paper was able to identify genes that code for lysosomal vacuolar ATPase (v-ATPase) that can lower levels of ataxin-2, a protein involved in ALS. Since several small molecule inhibitors of v-ATPase are already approved to treat other conditions, this is likely to yield positive results for ALS patients within relatively short timeframes.
A streamlined CRISPR workflow to introduce mutations and generate isogenic iPSCs for modeling amyotrophic lateral sclerosis.
CRISPR ALS mutations introduced to human induced pluripotent stem cellsA notable recent study provided a streamlined workflow for CRISPR editing in human induced pluripotent stem cells (iPSCs), introducing ALS genetic mutations such as those in SOD1 and FUS, as well as correcting them. Edited iPSCs can be then differentiated into clinically relevant motor neuron cells to provide accurate disease models.
A notable recent study provided a streamlined workflow for CRISPR editing in human induced pluripotent stem cells (iPSCs), introducing ALS genetic mutations such as those in SOD1 and FUS, as well as correcting them. Edited iPSCs can be then differentiated into clinically relevant motor neuron cells to provide accurate disease models.
CRISPR creates a TARDBP knock-in mouse model of ALSA CRISPR mouse model of ALS was generated in 2018, by knocking in the human ALS mutation to the mouse TARDBP gene. The resulting TDP-43Q331K mice were used to characterize the role of the mutation in ALS and how it affects the expression of other neurodegeneration-associated genes.
A CRISPR mouse model of ALS was generated in 2018, by knocking in the human ALS mutation to the mouse TARDBP gene. The resulting TDP-43Q331K mice were used to characterize the role of the mutation in ALS and how it affects the expression of other neurodegeneration-associated genes.
Inducing and correcting TARDBP mutations in human induced pluripotent stem cellsA 2019 CRISPR ALS study used human iPSCs to study the mechanisms by which TARDBP mutations and aberrant TDP-43 cause ALS. The authors found that TDP-43 malfunction impairs the secretion of brain-derived neurotrophic factor (BDNF) via altered splicing of Sortilin, a trafficking receptor. They also corrected the same mutation in patient-derived iPSCs.
A 2019 CRISPR ALS study used human iPSCs to study the mechanisms by which TARDBP mutations and aberrant TDP-43 cause ALS. The authors found that TDP-43 malfunction impairs the secretion of brain-derived neurotrophic factor (BDNF) via altered splicing of Sortilin, a trafficking receptor. They also corrected the same mutation in patient-derived iPSCs.
CRISPR ALS therapiesCRISPR has enormous potential to provide curative therapies by treating the underlying cause of genetic disease. It has now been used in many fALS studies to demonstrate that the genetic mutations that cause the disease can be either corrected to restore normal function or knocked out to eliminate the production of toxic proteins. Let’s explore some of the breakthrough CRISPR ALS research papers that have used these approaches to create novel gene therapies.
CRISPR has enormous potential to provide curative therapies by treating the underlying cause of genetic disease. It has now been used in many fALS studies to demonstrate that the genetic mutations that cause the disease can be either corrected to restore normal function or knocked out to eliminate the production of toxic proteins. Let’s explore some of the breakthrough CRISPR ALS research papers that have used these approaches to create novel gene therapies.
The History of CRISPR Cell and Gene Therapies
CRISPR cell and gene therapies can treat genetic diseases, cancer, and more. This article explores the history of cell and gene therapies, the CRISPR revolution, and the current state of the field.

CRISPR-Cas9 excision of the C9orf72 hyperexpansionA proof-of-concept Nature study published in 2022 demonstrated that the GGGGCC hyperexpansion in C9orf72 can be excised using CRISPR-Cas9. The authors employed an AAV vector to deliver the editing components to primary cortical neurons of ALS mice in vitro, and then to the brains of the same mouse models in vivo. They also tested their strategy in patient iPSC-derived motor neurons and brain organoids, rescuing disease phenotypes.
A proof-of-concept Nature study published in 2022 demonstrated that the GGGGCC hyperexpansion in C9orf72 can be excised using CRISPR-Cas9. The authors employed an AAV vector to deliver the editing components to primary cortical neurons of ALS mice in vitro, and then to the brains of the same mouse models in vivo. They also tested their strategy in patient iPSC-derived motor neurons and brain organoids, rescuing disease phenotypes.
CRISPR-Cas9 deletion of mutated SOD1CRISPR-Cas9 has shown promise in deleting mutated SOD1 in a mouse model of ALS. A 2020 Nature paper used the smaller Cas9 variant from Staphylococcus aureus (SaCas9) and delivered it to the mice via an AAV vector along with an sgRNA targeting the gene. This strategy was able to delete the gene, improving the lifespan of the mice by over 50%.
Another CRISPR ALS study used a similar method in the same mouse model, reporting that mutant SOD1 protein was reduced significantly in the spinal cord of the mice. The mice displayed improved motor function, reduced muscle atrophy, delayed disease onset, and increased survival.
CRISPR-Cas9 has shown promise in deleting mutated SOD1 in a mouse model of ALS. A 2020 Nature paper used the smaller Cas9 variant from Staphylococcus aureus (SaCas9) and delivered it to the mice via an AAV vector along with an sgRNA targeting the gene. This strategy was able to delete the gene, improving the lifespan of the mice by over 50%.
Another CRISPR ALS study used a similar method in the same mouse model, reporting that mutant SOD1 protein was reduced significantly in the spinal cord of the mice. The mice displayed improved motor function, reduced muscle atrophy, delayed disease onset, and increased survival.
CRISPR base editing to disable expression of SOD1CRISPR base editing systems have also been used to knock out the mutated SOD1 gene. The authors of a 2020 Molecular Therapy study employed an intein-mediated trans-splicing system to deliver cytidine base editors (CBEs) to a mouse model of ALS, introducing a nonsense mutation to knock out the mutant gene. Treated mice showed reduced muscle atrophy and increased neuromuscular function.
CRISPR base editing systems have also been used to knock out the mutated SOD1 gene. The authors of a 2020 Molecular Therapy study employed an intein-mediated trans-splicing system to deliver cytidine base editors (CBEs) to a mouse model of ALS, introducing a nonsense mutation to knock out the mutant gene. Treated mice showed reduced muscle atrophy and increased neuromuscular function.
CRISPR-based correction of mutated FUS gene in patient iPSCsIt is also possible to use CRISPR-Cas9 to correct ALS mutations, such as those in the FUS gene. A 2018 Nature Communications article demonstrated this approach in patient iPSC-derived motor neurons, reporting that DNA ligation defects in the cells that contribute to neurodegeneration can be rescued by CRISPR-mediated knock-in to correct the mutation.
It is also possible to use CRISPR-Cas9 to correct ALS mutations, such as those in the FUS gene. A 2018 Nature Communications article demonstrated this approach in patient iPSC-derived motor neurons, reporting that DNA ligation defects in the cells that contribute to neurodegeneration can be rescued by CRISPR-mediated knock-in to correct the mutation.
Hope for the Future: Can CRISPR Cure ALS?
Despite so many major advances in this field, CRISPR ALS research is still in its infancy, primarily because of the complex nature of this disease. Unlike monogenic disorders, which are caused by mutations in a single gene, fALS can be caused by a variety of different mutations, and the majority of ALS cases are not caused by inherited mutations at all.
Several companies are currently developing in vivo gene therapies for fALS using CRISPR technology, as well as further improving ALS disease models. Locana Bio, for example, is producing a CRISPR ALS therapy to delete mutated C9orf72 and is reportedly in the lead optimization phase of research. ixCells Biotechnologies are producing CRISPR-edited iPSCs for ALS disease modeling, including with SOD1 and TARDBP mutations.
In 2021, Capsida Biotherapeutics announced a partnership with CRISPR Therapeutics to develop and commercialize in vivo CRISPR gene therapies for amyotrophic lateral sclerosis and Friedreich’s ataxia, another neurodegenerative disease. Capsida is focussing on AAV capsid engineering in order to increase the transduction of brain cells in vivo and reduce the accumulation of AAVs in other organs. This strategy makes it possible to use lower doses of the viral vectors, which increases the safety profile of these therapies.
Scribe Therapeutics, a biotechnology company co-founded by CRISPR inventor Dr. Jennifer Doudna, is also taking aim at ALS. In 2020, Scribe announced a collaboration with fellow biotech company Biogen to develop in vivo CRISPR gene therapies for ALS.
Interestingly, although only 10% of ALS cases are caused by inherited genetic mutations, CRISPR may be able to help an even wider patient population. An increasing number of studies have demonstrated that a potential majority of ALS patients develop altered protein functions that contribute to the disease even without genetic mutations. This indicates that CRISPR technology could be used to switch off or otherwise alter the production of these problematic proteins - for example, using CRISPR inhibition (CRISPRi) or other methods.
Thanks to CRISPR technology and the concerted effort of scientists across the globe, we are beginning to unravel the complexity of ALS through improved disease models, and several potentially curative therapies have been demonstrated in proof-of-concept studies. Along with the potential for CRISPR to treat even the patients who do not carry genetic mutations, the future is looking considerably brighter for the ALS patient community and their families.