Gene Editing Treatments
CRISPR gene editing technology is widely popular for its potential to cure diseases. One of the most widely discussed examples in this regard is sickle cell anemia, a devastating blood disorder. Until recently, bone marrow transplant was the only real treatment for afflicted patients, but CRISPR gene therapy has ushered in new hope.
With the recent news about patients with sickle cell anemia being successfully treated using a CRISPR-based therapy, it has now become more of a reality. Learn all about ongoing research on various treatment options, and the current sickle cell disease gene therapy clinical trial results.
In this comprehensive guide, we cover everything you need to know about the world of sickle cell anemia.
Specifically, we will cover the following topics:
Sickle cell anemia is a well-known disease, affecting millions of people worldwide, and is the most common blood disorder in the United States. Identified around 100 years ago, sickle cell anemia was the first disease that was proven to have a molecular cause and has been widely researched. In this section, we’ll cover the basics of sickle cell anemia, including the cause of this condition and how it is inherited.
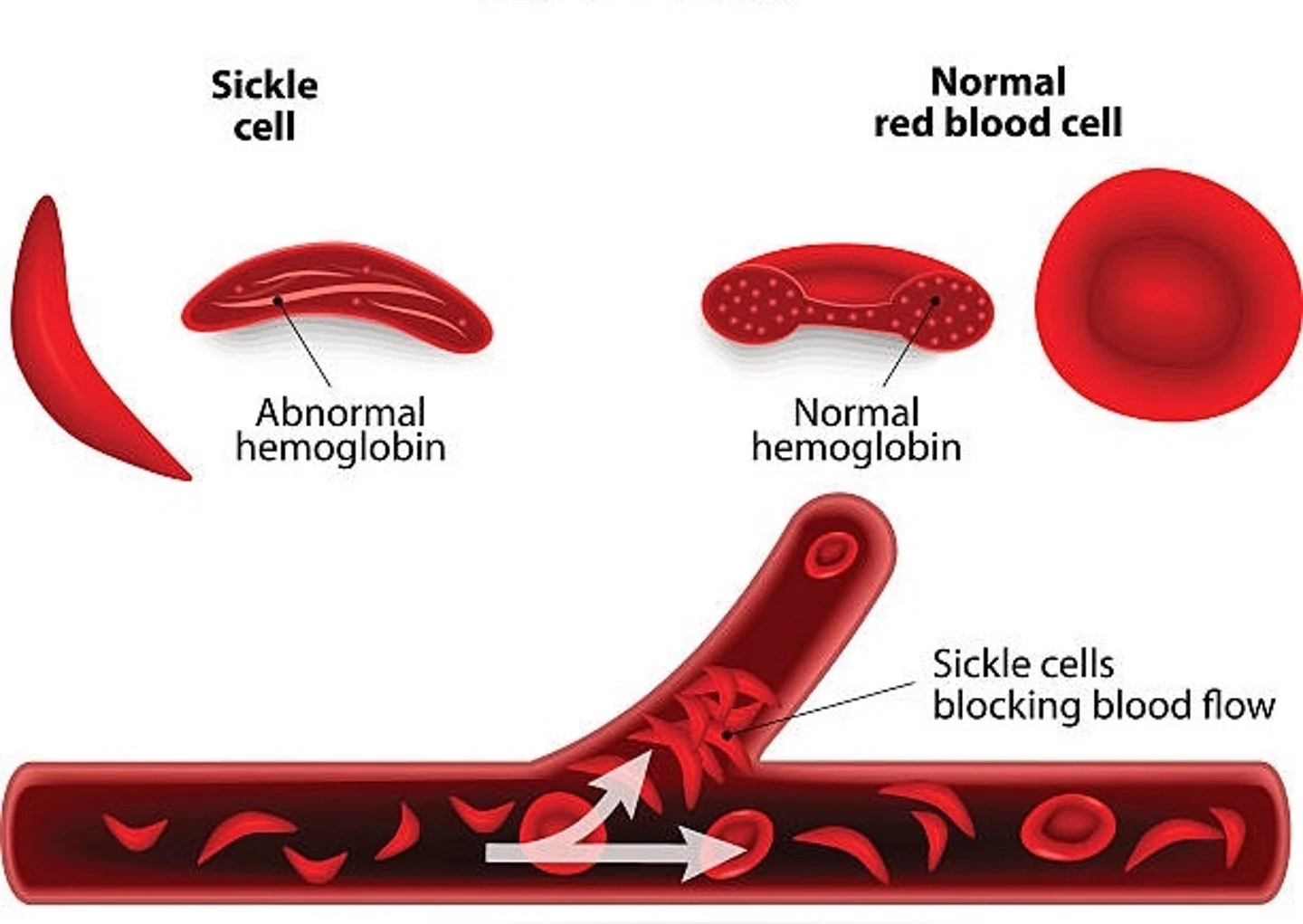
Sickle cell anemia is a genetic blood disorder that affects hemoglobin, the oxygen-transporting molecule in red blood cells. Sickle cell disease causes the body to produce hemoglobin S, an abnormal form of the molecule that distorts the shape of red blood cells (resembling a sickle), disrupting their function.
Individuals affiliated with this disorder produce deoxygenated hemoglobin S molecules, which cause red blood cells to distort and form a rigid, crescent (sickle) shape. The affected cells have a reduced lifespan in the body, resulting in shortages of red blood cells. The rigid, distorted cells obstruct blood vessels and inhibit circulation, causing severe pain and recurrent infections. Obstructions often result in clogs in the spleen, liver, lungs, heart, or eyes. Sickle cell disease causes increasing organ and tissue damage over time and increases the risk of stroke.
Sickle cell anemia is caused by the mutation of a single base in the DNA sequence of the ß-globin gene, HBB. In healthy individuals, position 6 of the resulting amino acid sequence is a glutamic acid (GAG), however, in sickle cell anemia patients, this is substituted for a valine (GTG). This mutation results in the formation of hemoglobin S, the disease-associated form of the protein.
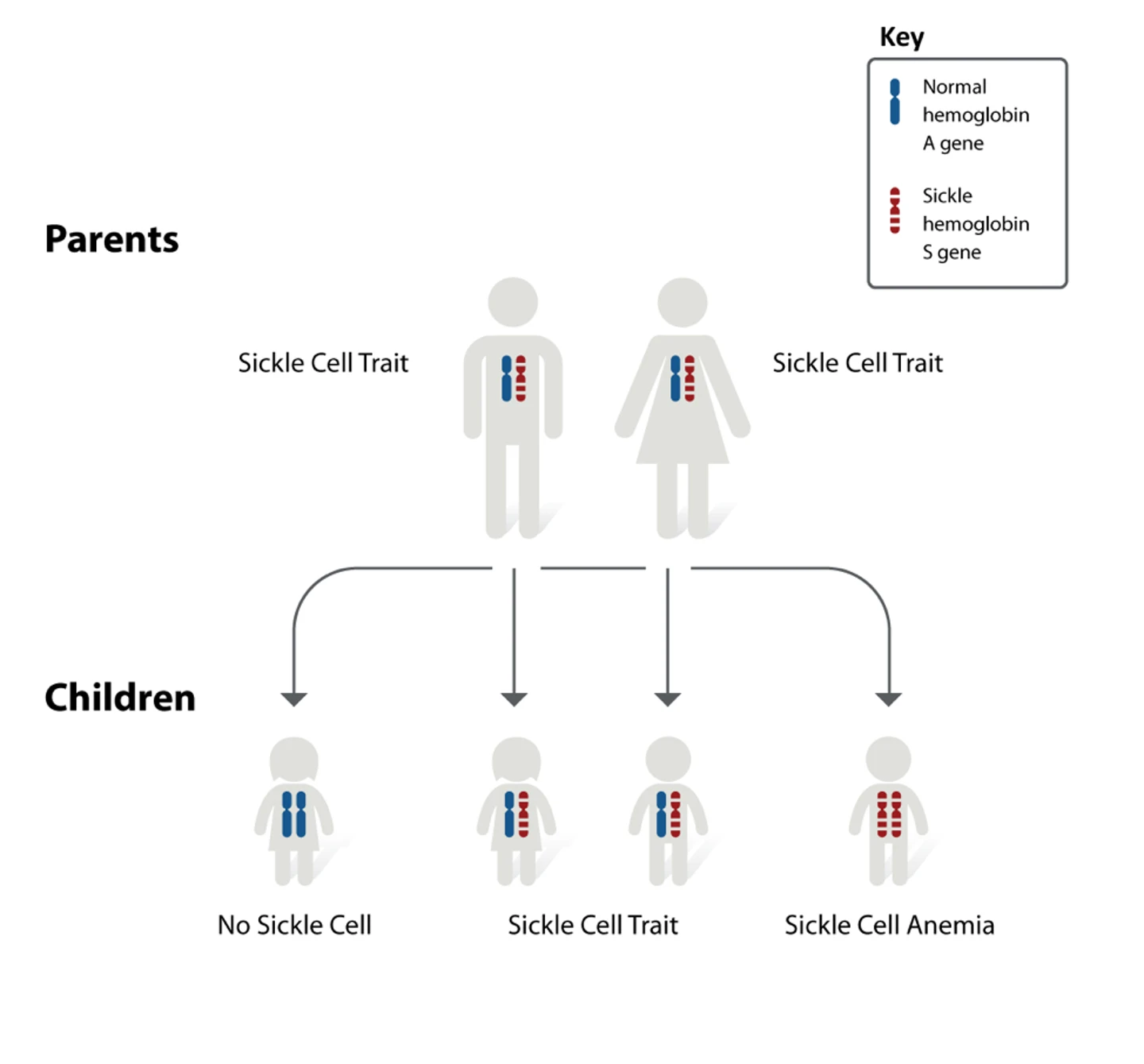
Sickle cell anemia is a monogenic, autosomal recessive condition. Monogenic means that it is caused by a mutation in a single gene, autosomal means that the gene is not located on a sex chromosome, and recessive means that an individual only develops the disease if the mutation is inherited from both parents.
Inheritance of only one copy causes sickle cell trait, which usually does not cause symptoms, but places individuals at risk of passing the mutation on to their offspring. Sickle cell anemia is also sometimes referred to as a Mendelian disease because of the way it is inherited, and as such, it is commonly used to explain genetics and Mendelian inheritance. The condition disproportionately affects certain ethnicities, particularly people of African-American heritage.
The difference between sickle cell anemia and beta-thalassemia is that sickle cell anemia is caused by a very specific mutation in the HBB gene, while beta-thalassemia can be caused by multiple mutations in the same gene. While they have similar outcomes, beta-thalassemia does not cause red blood cells to sickle.
It is also possible for individuals to have both types of mutation, resulting in a condition known as sickle beta-thalassemia. For details on beta-thalassemia, visit our CRISPR in beta-thalassemia page.
Sickle cell disease is diagnosed via blood test, however, CRISPR-Cas9 systems have now been adapted as more rapid diagnostic tools for the genetic mutation that causes the disease. One recently developed CRISPR diagnostic tool for sickle cell anemia is the SNP-Chip (see link below), which uses CRISPR to detect single nucleotide polymorphisms, like the one that causes sickle cell anemia.
Treatment options for sickle cell anemia are currently limited. Patients typically require frequent blood transfusions, and medications are typically recommended to relieve the pain attacks or reduce their frequency. Some medications can promote fetal hemoglobin (hbF) production to replace the faulty adult hemoglobin, however, they carry associated health risks. Antibiotics are often prescribed to patients in order to prevent frequent bacterial infections, particularly in children.
A common cure available for sickle cell disease is the transplantation of bone marrow from a healthy donor. This approach presents significant challenges, including identifying a suitable donor, immune rejection of the transplant, and graft-versus-host disease (GVHD). It is also typically restricted to children and young people because the associated risks increase with the age of the patient.
More recently in December of 2023, the FDA approved the first CRISPR-based therapy, called Casgevy. Developed by CRISPR Therapeutics and Vertex Pharmaceuticals, Casgevy specifically targets the BCL11A gene to treat sickle cell disease and beta-thalassemia. As you continue, we will discuss the impact CRISPR has in treating sickle cell disease, Casgevy, and other CRISPR-based therapeutics scientists have developed.
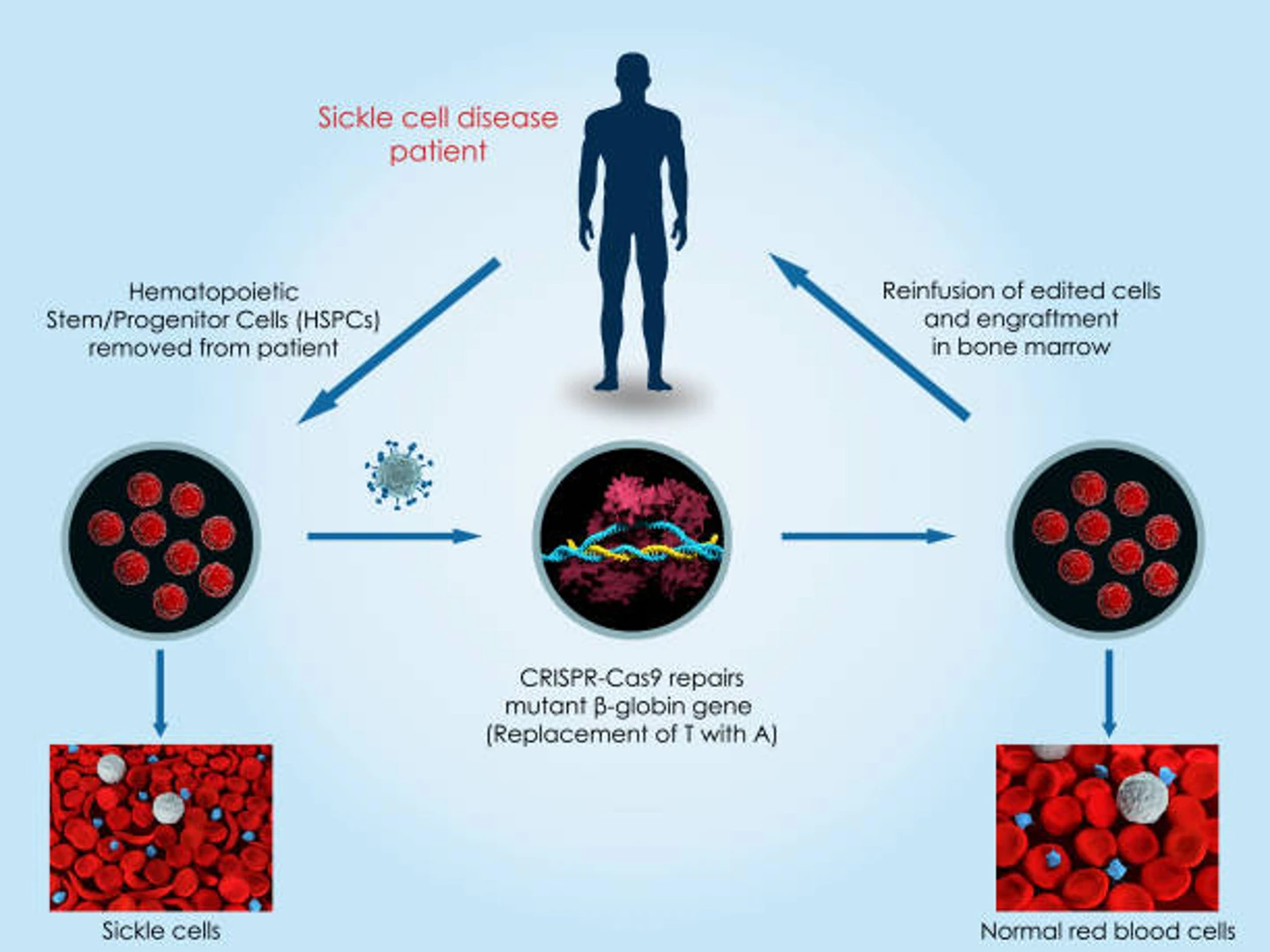
As sickle cell disease is caused by a genetic mutation, it is a perfect candidate for CRISPR-mediated gene therapy. Treating sickle cell anemia with CRISPR involves an ex vivo procedure known as gene-edited cell therapy, where hematopoietic stem cells are extracted from the patient, corrected, and then replaced. In this section, we’ll discuss the main approaches scientists are using to create CRISPR sickle cell gene-edited cell therapies.
One of the main approaches to CRISPR sickle cell gene therapy is to repair the mutation in the adult hemoglobin gene responsible for sickle cell disease, causing the healthy, normal form of adult hemoglobin (hemoglobin S) to be produced. Daniel Dever, a research instructor in Matthew Porteus' lab at Stanford University uses this approach in his work involving CRISPR to introduce a DNA break to the ß-globin gene.
The site of the break can then be used to introduce a correction to the gene via homology-directed repair (HDR). This is called a gene knock-in; a donor template containing the normal sequence of the gene is introduced so that the mutation is corrected when the cell repairs the DNA break with the template via HDR. The edited cells, now engineered to produce normal hemoglobin, are re-implanted in the patient’s bloodstream.
The preclinical development of Dr. Dever’s ß-globin gene-editing method is complete, and the method proves to be incredibly efficient and reproducible, allowing for mutation and analysis of precise genomic locations in weeks. Clinical trials are the next steps toward the implementation of this treatment.
The second approach to CRISPR sickle cell gene therapy involves a gene knockout, switching off the gene that suppresses fetal hemoglobin. This method causes fetal hemoglobin (hemoglobin F) to be expressed, replacing the mutated adult hemoglobin.
This is a highly promising CRISPR-Cas9 sickle cell therapy - by mutating (knocking out) the BCL11A gene, the expression of hemoglobin F is indirectly promoted. This approach was developed after it was found that sickle cell patients with a natural mutation in their BCL11A gene were resistant to disease symptoms.
CTX001, also referred to Casgevy, is one such investigational new drug application, co-sponsored by CRISPR Therapeutics and Vertex Pharmaceuticals, to treat sickle cell disease. The drug has successfully corrected sickle cell disease in transgenic mice with clinically relevant efficiency. In the next section, we will discuss further the successful results from patients who participated in a CTX001 clinical trial.
Outside of Casgevy, which was approved by the FDA and EMA for clinical applications, there are several other possibilities for CRISPR sickle cell therapies. The first, and perhaps most feasible, is through the use of CRISPR base editors or prime editors. These highly precise CRISPR systems can induce point mutations without causing double-stranded breaks in DNA and are therefore considered a safer alternative to traditional CRISPR editing.
Base editing has been shown to successfully alleviate sickle cell anemia in vivo in mice, however, the authors took an unusual approach to the problem. Because base editing systems are not capable of inducing all possible point mutations, they cannot be used to change the mutated GTG codon found in sickle cell anemia patients to the GAG that is present in healthy individuals. Instead, the authors changed the thymine nucleotide to a cytosine, resulting in a GCG codon. This change results in a non-pathogenic variant of HBB known as Makassar β-globin (HBBG), which functions like healthy adult β-globin.
Another CRISPR-based approach to treating sickle cell anemia is epigenetic editing. A 2017 study found that fetal hemoglobin expression is mediated by epigenetic factors, with a genetic variant delaying the switch from fetal hemoglobin to the adult form. Based on this finding, other researchers have speculated that CRISPR epigenetic editing could be used to upregulate fetal hemoglobin in sickle cell anemia patients. This approach would not result in permanent, heritable changes in the genome of patients, and therefore may be a viable alternative to traditional CRISPR editing.
CRISPR technology had several key breakthroughs in 2020, one of them being the first successful clinical trial treating sickle cell anemia. In this section, we’ll take a closer look at some of the clinical trials for CRISPR sickle cell disease gene therapies.
One of the most media-covered clinical trials was CTX001 (Casgevy), an autologous gene-edited cell therapy developed by CRISPR Therapeutics and Vertex Pharmaceuticals. This CRISPR cell therapy clinical trial for treating sickle cell disease involves restoring the expression of fetal hemoglobin to alleviate the symptoms of sickle cell disease and beta-thalassemia. Bone marrow stem cells are extracted from patients and CRISPR-edited to inactivate BCL11A, a repressor of fetal hemoglobin production. The edited stem cells are introduced back into the patient with the hope that the new red blood cells produced by the edited stem cells would have fetal hemoglobin.
Victoria Gray was the first CRISPR sickle cell patient, having fought the disease for 34 years. Following the one-time treatment, her blood showed a significant proportion of fetal hemoglobin levels, and she has been able to avoid blood transfusions and pain attacks without any major side effects. Dr. Haydar Frangoul, the medical director of pediatric hematology/oncology at HCA Healthcare’s Sarah Cannon Research Institute Center, who has been treating Gray, said the following, as per the article.
“She is functioning as somebody who does not have sickle cell disease. I believe this is absolutely, totally transformative therapy” -Dr. Haydar Frangoul.
In the American Society of Hematology annual meeting 2020, the data presented showed that a total of 10 patients—three with sickle cell disease and seven with beta-thalassemia, have shown tremendous progress. An updated report published in December 2020 in NEJM showed effective and safe Casgevy therapy results in 2 sickle cell anemia and beta-thalassemia patients after 15 months and 18 months, respectively.
In June of 2021, the researchers presented more long-term results of 22 patients at the European Hematology Association Annual Meeting, demonstrating consistent and sustained responses from the patients following treatment. With such promising data, this treatment approach is looking more promising to become mainstream.
After the positive results from clinical trials, the U.S. Food and Drug Administration (FDA) approved Csgevy for patients aged 12 years or older in December of 2023. Shortly after the FDAs decision, the European Medicines Agency (EMA). With its approval, individuals who have sickle disease have an option that can cure the disease improving their quality of life. With Casgevy’s approval, we sat down with Jimi Olaghere, one of the first patients to receive Casgevy during clinical trials, to discuss his perspective on the landmark approval of the therapy to treat sickle cell disease. We also sat down with our Associate Director of Regulatory Affairs, Lina Jamis, to discuss the approval of Casgevy’s impact on the regulatory landscape.
The RUBY trial, spearheaded by Editas, is a phase I/II trial of an autologous CD34+ hematopoietic stem cell therapy called EDIT-301. This treatment takes the same approach as Casgevy, extracting patient cells, editing them ex vivo to upregulate fetal hemoglobin, and reintroducing them to the patients.
EDIT-301 utilizes the Cas12a nuclease to edit the hemoglobin subunit gamma 1 and 2 (HBG1/2) promoter region to increase fetal hemoglobin production. Early results from patients in the EDIT-301 clinical trials showed successful engrafting and increases in their hemoglobin levels. Editas Medicine is also investigating if EDIT-301 therapy could also treat transfusion-dependent beta-thalassemia (TDT).

Graphite Bio is taking a different approach to treating sickle cell anemia, hoping to cure the condition by restoring the expression of adult hemoglobin rather than replacing it with fetal hemoglobin. The GPH101 treatment, developed by Stanford University and Graphite Bio co-founder Matthew Porteus, is an autologous CD34+ HSPC transplant.
GPH101 uses gene editing to correct the mutation present in the HBB gene of sickle cell disease patients and restore normal hemoglobin expression. In early 2023, Graphite Bio voluntarily paused GPH101 the phase I/II clinical trials because patients developed adverse side effects. Although possibly not linked directly to GPH101, the pause was decided to protect the patient's health and safety.
| Trial | Treatment name | Editing method/strategy | Company or clinician | Status |
|---|---|---|---|---|
| NCT03745287 | CTX001 (Casgevy) | Restored fetal hemoglobin by knocking out BCL11A transcription factor in autologous CD34+ HSPCs via CRISPR-Cas9 | Vertex Pharmaceuticals Inc. CRISPR Therapeutics | Complete, successful |
| NCT04443907 | OTQ923 and HIX763 | Autologous HSPC transplant; gene disruption to restore fetal hemoglobin via CRISPR-Cas9 | Novartis Pharmaceuticals | Phase I/II |
| NCT04925206 | ET-01 | Autologous HSPC transplant; gene disruption to restore fetal hemoglobin (Beta-thalassemia) via CRISPR-Cas9 | EdiGene, Inc. | Phase I |
| NCT04853576 (RUBY Trial) | EDIT-301 | Autologous CD34+ HSPC transplant, editing the Haemoglobin Subunit Gamma 1 and 2 (HBG1/2) promoter region to increase fetal hemoglobin production via CRISPR-Cas12a | Editas Medicine, Inc. | Phase I/II |
| NCT04774536 | CRISPR_SCD001 | Autologous HSPC transplant, replacing mutated beta-globin gene via CRISPR-Cas9 knock-in using non-viral delivery methods | UCLA, UC Berkeley | Phase I/II |
| NCT04819841 (CEDAR Trial) | GPH101 | Autologous CD34+ HSPC transplant, correcting the mutation in HBB to restore normal hemoglobin expression | Graphite Bio, Inc. | Phase I/II |
During World CRISPR Day 2020, we had the pleasure of hosting Dr. Matthew Porteus at Stanford and Dr. Donald Kohn at UCLA to speak about the therapies they developed. To learn more, watch the recording below.
We also had the pleasure of hosting Mark DeWitt and Don Kohn on the CRISPR Cuts podcast, where they spoke deeper into how their therapy was developed to treat sickle cell disease. They also emphasized the value of using one company as their CRISPR reagent supplier to support their work as a continuum from research use only (RUO) gRNA to good manufacturing practice (GMP) grade gRNA, as it helped streamline their therapeutic development.
In 2021, the, the FDA has approved a new clinical trial to treat the root cause of sickle cell anemia. Spearheaded by Dr. Mark Walters, professor of pediatrics at UCSF, the autologous cell therapy trial aims to directly target and correct the disease-causing mutation via gene knock-in. The researchers predict that editing efficiencies of 20% should be enough to result in significant clinical benefit to sickle cell patients.
Beam Therapeutics has also set its sights on treating sickle cell disease via a base editing approach and received FDA clearance to begin clinical trials. The treatment, known as BEAM-101, is an autologous hematopoietic cell therapy that introduces point mutations known to occur in people who maintain the expression of fetal hemoglobin. Beam Therapeutics is also in the process of gathering preclinical data for another base editing treatment, BEAM-102, which corrects the disease-causing mutation in the adult hemoglobin gene. In 2022, BEAM Therapeutics submitted their IND for BEAM-102.
As current CRISPR-based therapies progress through the clinic and new therapies are developed, assessing their safety and efficacy is crucial as this groundbreaking technology continues to revolutionize the landscape of medical treatment. Rigorous evaluation ensures that these innovative approaches do not pose unintended risks while maximizing their therapeutic potential. As CRISPR offers unprecedented precision in gene editing, it is reshaping how therapies are developed, enabling scientists to target and potentially cure genetic disorders with remarkable accuracy.
Furthermore, scientists are actively exploring novel nucleases as alternatives because they lower the entry barrier to developing therapies because of their improved licensing structures and gene editing versatility. These novel nucleases, including hfCas12Max and eSpOT-ON (as recombinant protein or mRNA), offer distinct advantages in terms of target specificity and reduced off-target effects, broadening the range of applications for CRISPR-based therapies. By integrating these alternative nucleases into their toolkit, scientists can refine existing therapies and develop innovative solutions for previously challenging gene targets. This evolution in the CRISPR landscape not only accelerates the therapeutic development process but also fosters personalized medicine, tailoring treatments to individual genetic profiles. As we move forward, ongoing research and regulatory scrutiny will be essential to harnessing the full potential of CRISPR, paving the way for a future where previously untreatable diseases may become manageable or even curable.