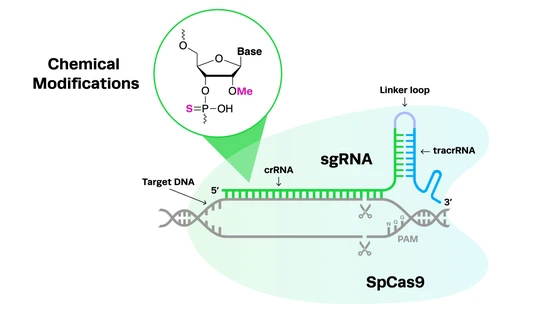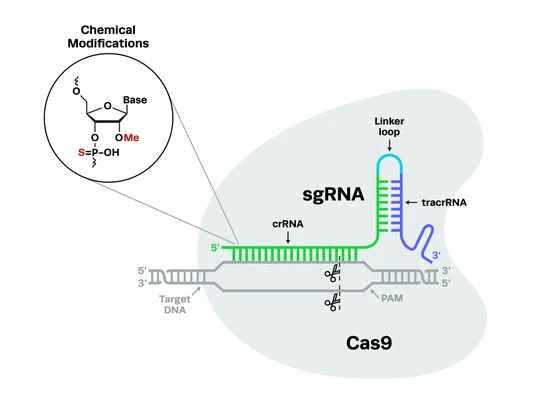
Complex genetic disorders like cystic fibrosis are desperately in need of more accurate disease models and novel therapies. While life expectancies for patients have increased significantly in recent decades, there is no functional cure for the disease, and a subset of patients have no available treatment options.
In this new era of genomic medicine, CRISPR has the potential to answer the unmet needs of the patient community. This blog is a deep dive into cystic fibrosis, from the underlying biology of this devastating disease, to how CRISPR can create better disease models and possible cures.
Cystic Fibrosis: Causes, Genetics, Symptoms, Diagnosis, and Treatment
Cystic fibrosis is a complex disease and can manifest very differently from one individual to another. Let’s take a look at the basics of cystic fibrosis, including the underlying cause of this disease, genetics and inheritance, symptoms, diagnosis, and current treatment options.
What is cystic fibrosis?Cystic fibrosis (CF) is a severe genetic disease that affects multiple organs, including the lungs and pancreas. It is progressive and can be fatal, primarily through respiratory failure, multi-organ dysfunction, and frequent infections. There is currently no cure for this disease.
Cystic fibrosis (CF) is a severe genetic disease that affects multiple organs, including the lungs and pancreas. It is progressive and can be fatal, primarily through respiratory failure, multi-organ dysfunction, and frequent infections. There is currently no cure for this disease.
What causes cystic fibrosis and how does it affect the body?Cystic fibrosis is caused by loss-of-function mutations in the cystic fibrosis transmembrane regulator (CFTR) gene, which codes for a chloride channel in the membrane of epithelial cells. These mutations mean the CFTR protein doesn’t do its job properly, there isn’t enough of it, or it isn’t produced at all.
The mutated CFTR protein results in impaired flow of salts through the channel, disrupting homeostasis in cells. Let’s take a lung epithelial cell as an example: when chloride ions cannot pass through the membrane to the outside of the cell, there is insufficient salt on the surface to attract water. Without water, the mucus layer on the cell surface becomes thick and sticky, and the cilia cannot do their job of sweeping back and forth to move this mucus throughout the airway.
The combination of thickened mucus and impaired function of the cilia means the mucus becomes stuck in the airways, causing significant breathing difficulties. The accumulation of thickened mucus is known as mucostasis. It also traps pathogens like bacteria and viruses in the airways, causing frequent infections that can be fatal. CF patients experience cycles of mucostasis, inflammation, and infection that result in progressive damage to the lungs.
Cystic fibrosis is often thought of as a disease of the lungs, but it also affects multiple other organs. In the pancreas, thickened mucus can build up and impair the release of digestive enzymes; without these enzymes, the body cannot properly absorb nutrients from food, leading to nutrient deficiencies and growth issues. Similarly, mucus can block the bile duct of the liver, causing buildup of bile and liver disease. It also affects the gastrointestinal system and can cause fertility issues in men.
Cystic fibrosis is caused by loss-of-function mutations in the cystic fibrosis transmembrane regulator (CFTR) gene, which codes for a chloride channel in the membrane of epithelial cells. These mutations mean the CFTR protein doesn’t do its job properly, there isn’t enough of it, or it isn’t produced at all.
The mutated CFTR protein results in impaired flow of salts through the channel, disrupting homeostasis in cells. Let’s take a lung epithelial cell as an example: when chloride ions cannot pass through the membrane to the outside of the cell, there is insufficient salt on the surface to attract water. Without water, the mucus layer on the cell surface becomes thick and sticky, and the cilia cannot do their job of sweeping back and forth to move this mucus throughout the airway.
The combination of thickened mucus and impaired function of the cilia means the mucus becomes stuck in the airways, causing significant breathing difficulties. The accumulation of thickened mucus is known as mucostasis. It also traps pathogens like bacteria and viruses in the airways, causing frequent infections that can be fatal. CF patients experience cycles of mucostasis, inflammation, and infection that result in progressive damage to the lungs.
Cystic fibrosis is often thought of as a disease of the lungs, but it also affects multiple other organs. In the pancreas, thickened mucus can build up and impair the release of digestive enzymes; without these enzymes, the body cannot properly absorb nutrients from food, leading to nutrient deficiencies and growth issues. Similarly, mucus can block the bile duct of the liver, causing buildup of bile and liver disease. It also affects the gastrointestinal system and can cause fertility issues in men.
Cystic fibrosis geneticsThere are thousands of different mutations that can occur in the CFTR gene, several hundred of which are known to cause cystic fibrosis. They are categorized into five classes based on how they affect the CFTR protein: protein production, insufficient protein, protein processing, gating, and conduction mutations.
These mutations are distributed along almost the entire CFTR gene, including in both introns and exons. ΔF508, a protein processing mutation, is the most common.
There are thousands of different mutations that can occur in the CFTR gene, several hundred of which are known to cause cystic fibrosis. They are categorized into five classes based on how they affect the CFTR protein: protein production, insufficient protein, protein processing, gating, and conduction mutations.
These mutations are distributed along almost the entire CFTR gene, including in both introns and exons. ΔF508, a protein processing mutation, is the most common.
Is cystic fibrosis dominant or recessive?Cystic fibrosis is a recessive disease. This means someone would need to inherit two copies of the mutated CFTR gene to develop the disease. If someone only inherits one copy of the mutated gene, they won’t develop the disease but they will be a carrier and can pass it on to their children.
Cystic fibrosis is a recessive disease. This means someone would need to inherit two copies of the mutated CFTR gene to develop the disease. If someone only inherits one copy of the mutated gene, they won’t develop the disease but they will be a carrier and can pass it on to their children.
How common is cystic fibrosis?Worldwide, around 70,000 people suffer from cystic fibrosis. According to the Cystic Fibrosis Foundation (CFF), almost 40,000 children and adults in the US currently live with this disease, and around 1,000 new cases are confirmed every year. Despite common misconceptions, it can affect people of any ethnicity.
Worldwide, around 70,000 people suffer from cystic fibrosis. According to the Cystic Fibrosis Foundation (CFF), almost 40,000 children and adults in the US currently live with this disease, and around 1,000 new cases are confirmed every year. Despite common misconceptions, it can affect people of any ethnicity.
Cystic fibrosis symptomsThere are a wide variety of symptoms of cystic fibrosis, however, the most common include a persistent cough, wheezing and shortness of breath, poor growth in children, digestive issues and problems gaining weight, frequent lung infections, nasal polyps and chronic sinus infections, and very salty sweat.
There are a wide variety of symptoms of cystic fibrosis, however, the most common include a persistent cough, wheezing and shortness of breath, poor growth in children, digestive issues and problems gaining weight, frequent lung infections, nasal polyps and chronic sinus infections, and very salty sweat.
Cystic fibrosis diagnosisDiagnosing cystic fibrosis typically requires multiple steps: a sweat test, a screen for CFTR mutations, and a clinical evaluation by a disease specialist. In the US, screening is performed to detect the disease in newborns, and as such, most people are diagnosed during very early childhood.
Diagnosing cystic fibrosis typically requires multiple steps: a sweat test, a screen for CFTR mutations, and a clinical evaluation by a disease specialist. In the US, screening is performed to detect the disease in newborns, and as such, most people are diagnosed during very early childhood.
Cystic fibrosis treatmentTreatment options for cystic fibrosis are limited. Around 90% of CFTR mutations can be targeted with small molecule drugs known as CFTR modulators, which can improve the trafficking and processing of the CFTR protein. However, around 10% of CFTR mutations have no available treatment options.
According to the CFF, a child born with the disease in recent years could live to around 56 years of age, a significant increase from previous predictions. While CFTR modulators have increased the life expectancy and quality of life of patients, they are not curative and they cannot reverse any organ damage caused by the disease.
Before CRISPR offered the possibility of gene correction, several clinical trials attempted to treat CF in the airways of patients using gene replacement therapy, in which a functional copy of the CFTR cDNA is delivered to cells. Unfortunately, these trials were unsuccessful, with no durable responses seen in the patients.
Treatment options for cystic fibrosis are limited. Around 90% of CFTR mutations can be targeted with small molecule drugs known as CFTR modulators, which can improve the trafficking and processing of the CFTR protein. However, around 10% of CFTR mutations have no available treatment options.
According to the CFF, a child born with the disease in recent years could live to around 56 years of age, a significant increase from previous predictions. While CFTR modulators have increased the life expectancy and quality of life of patients, they are not curative and they cannot reverse any organ damage caused by the disease.
Before CRISPR offered the possibility of gene correction, several clinical trials attempted to treat CF in the airways of patients using gene replacement therapy, in which a functional copy of the CFTR cDNA is delivered to cells. Unfortunately, these trials were unsuccessful, with no durable responses seen in the patients.
Understanding CRISPR Clinical Trials: Your Questions Answered
CRISPR gene therapies are the future of medicine! But what does it take to develop a CRISPR drug? How do CRISPR clinical trials work, and how does the FDA approve them? In this blog, we answer all your questions about CRISPR clinical trials.

Can CRISPR Cure Cystic Fibrosis?
CRISPR has the potential to offer one-time curative therapies for genetic diseases. So how can CRISPR be used to treat cystic fibrosis? Let’s explore the unique challenges of developing a CRISPR treatment for cystic fibrosis, the ways CRISPR has improved CF disease models, ex vivo and in vivo approaches to CRISPR cystic fibrosis therapy, and the potential for CRISPR cystic fibrosis clinical trials.
Challenges in creating a CRISPR treatment for cystic fibrosisAs we mentioned earlier, there are around 1,700 different mutations in CFTR that can cause cystic fibrosis, and they are distributed throughout the entire gene. This alone presents a major challenge when developing CRISPR-based therapies for the disease - to treat all CF patients, researchers would need to correct all of these mutations, essentially replacing the entire gene.
This problem is exacerbated by the sheer size of the CFTR gene - the coding region alone is 4.5kb, making it difficult to deliver in vivo to cells and tissues. For example, adeno-associated viral vectors (AAVs), which are frequently used to deliver gene therapies in vivo, have a packaging limit of around 4.7kb. This makes it impossible to deliver the CFTR template, gene editing components like Cas9, and a single guide RNA in a single AAV vector.
While it is more feasible to deliver these gene-editing components to tissues in vivo by packaging them in a lipid nanoparticle (LNP), generating LNPs that can target organs other than the liver is another obstacle to the successful treatment of the disease.
Moreover, the airway epithelium is primarily made up of non-dividing cells, preventing efficient correction of genetic mutations via homology-directed repair (HDR). To successfully treat the disease with in vivo CRISPR editing, the airway stem and progenitor cells would need to be specifically targeted.
In contrast, developing ex vivo CRISPR-edited cell therapies for cystic fibrosis presents other challenges, including difficulties in the transplant and engraftment of edited cells.
As we mentioned earlier, there are around 1,700 different mutations in CFTR that can cause cystic fibrosis, and they are distributed throughout the entire gene. This alone presents a major challenge when developing CRISPR-based therapies for the disease - to treat all CF patients, researchers would need to correct all of these mutations, essentially replacing the entire gene.
This problem is exacerbated by the sheer size of the CFTR gene - the coding region alone is 4.5kb, making it difficult to deliver in vivo to cells and tissues. For example, adeno-associated viral vectors (AAVs), which are frequently used to deliver gene therapies in vivo, have a packaging limit of around 4.7kb. This makes it impossible to deliver the CFTR template, gene editing components like Cas9, and a single guide RNA in a single AAV vector.
While it is more feasible to deliver these gene-editing components to tissues in vivo by packaging them in a lipid nanoparticle (LNP), generating LNPs that can target organs other than the liver is another obstacle to the successful treatment of the disease.
Moreover, the airway epithelium is primarily made up of non-dividing cells, preventing efficient correction of genetic mutations via homology-directed repair (HDR). To successfully treat the disease with in vivo CRISPR editing, the airway stem and progenitor cells would need to be specifically targeted.
In contrast, developing ex vivo CRISPR-edited cell therapies for cystic fibrosis presents other challenges, including difficulties in the transplant and engraftment of edited cells.
Cystic fibrosis CRISPR disease modelsTo better understand cystic fibrosis and generate new treatments, scientists need to model the disease in animals. Yet researchers have long struggled to generate animal models that can accurately recapitulate the disease phenotype, including the airway bacterial infections commonly experienced by CF patients. This is largely because mice – the most common small animal model used in preclinical research – differ considerably from humans in the biology of their airways.
CRISPR editing has contributed to a recent breakthrough in cystic fibrosis disease modeling, generating genetically modified ferrets. With more similarities to human lung biology, ferrets were used to study pulmonary ionocytes, a subtype of airway epithelial cells that highly express the CFTR protein.
After editing the CFTR gene in ferret embryos, researchers investigated the role of pulmonary ionocytes in the regulation of airway fluids. Another recent study used primary stem cells from adult patients to generate intestinal organoids for disease modeling, demonstrating the repair of CFTR mutations. Other researchers are investigating the regulation of CFTR using CRISPR epigenetic editing modalities.
To better understand cystic fibrosis and generate new treatments, scientists need to model the disease in animals. Yet researchers have long struggled to generate animal models that can accurately recapitulate the disease phenotype, including the airway bacterial infections commonly experienced by CF patients. This is largely because mice – the most common small animal model used in preclinical research – differ considerably from humans in the biology of their airways.
CRISPR editing has contributed to a recent breakthrough in cystic fibrosis disease modeling, generating genetically modified ferrets. With more similarities to human lung biology, ferrets were used to study pulmonary ionocytes, a subtype of airway epithelial cells that highly express the CFTR protein.
After editing the CFTR gene in ferret embryos, researchers investigated the role of pulmonary ionocytes in the regulation of airway fluids. Another recent study used primary stem cells from adult patients to generate intestinal organoids for disease modeling, demonstrating the repair of CFTR mutations. Other researchers are investigating the regulation of CFTR using CRISPR epigenetic editing modalities.
Advances in CRISPR Disease Modeling: Cells, Organoids, Animals, and More
Disease modeling helps us understand disease and create therapies. This blog covers types of disease models, the impact of CRISPR, current challenges, and trends.

Cystic fibrosis CRISPR ex vivo cell therapyOne potential avenue to treating cystic fibrosis with CRISPR is through ex vivo cell therapy. Two independent studies published in 2020 used CRISPR-Cas9 to correct mutations in CFTR. In one paper, a team of researchers from Stanford University led by Matthew Porteus demonstrated the correction of the ΔF508 mutation in patient-derived basal airway stem cells using a Cas9 ribonucleoprotein (RNP).
The team was able to show that the correction of just 30% of ΔF508 alleles was sufficient to restore CFTR function in the cells, and that edited cells were able to differentiate after embedding in a scaffold. The other study, published in Molecular Therapy, showed the correction of the same ΔF508 mutation as well as the integration of a partial CFTR cDNA template to correct multiple mutations at once.
In 2022, the Stanford team developed a method to correct all CFTR mutations in cells ex vivo, overcoming delivery limitations by splitting the CFTR repair template and gene-editing components across two AAV vectors. They corrected the gene in both upper airway basal stem cells (UABCs) and human bronchial epithelial cells (HBECs), demonstrating 70% restored CFTR function in cultured cells.
One potential avenue to treating cystic fibrosis with CRISPR is through ex vivo cell therapy. Two independent studies published in 2020 used CRISPR-Cas9 to correct mutations in CFTR. In one paper, a team of researchers from Stanford University led by Matthew Porteus demonstrated the correction of the ΔF508 mutation in patient-derived basal airway stem cells using a Cas9 ribonucleoprotein (RNP).
The team was able to show that the correction of just 30% of ΔF508 alleles was sufficient to restore CFTR function in the cells, and that edited cells were able to differentiate after embedding in a scaffold. The other study, published in Molecular Therapy, showed the correction of the same ΔF508 mutation as well as the integration of a partial CFTR cDNA template to correct multiple mutations at once.
In 2022, the Stanford team developed a method to correct all CFTR mutations in cells ex vivo, overcoming delivery limitations by splitting the CFTR repair template and gene-editing components across two AAV vectors. They corrected the gene in both upper airway basal stem cells (UABCs) and human bronchial epithelial cells (HBECs), demonstrating 70% restored CFTR function in cultured cells.
CRISPR cystic fibrosis in vivo gene therapyAnother approach to creating a CRISPR treatment for cystic fibrosis is in vivo gene therapy, where cells and tissues are edited inside the body. In February 2024, Intellia Therapeutics and ReCode Therapeutics announced a major strategic collaboration to create life-changing therapies for the disease using this method.
With an initial focus on developing therapies for patients who cannot be treated with CFTR modulators, the partners will combine Intellia’s CRISPR-Cas9 DNA writing technology with ReCode’s selective organ targeting (SORT) LNPs to correct CFTR mutations in the lungs of patients via HDR.
A Nature Communications study published in 2023 by scientists from the University of Texas Southwestern Medical Center demonstrated proof-of-concept of this approach in mice. If their initial approach is successful, Intellia and ReCode plan to expand their program to target other CFTR mutations.
Another approach to creating a CRISPR treatment for cystic fibrosis is in vivo gene therapy, where cells and tissues are edited inside the body. In February 2024, Intellia Therapeutics and ReCode Therapeutics announced a major strategic collaboration to create life-changing therapies for the disease using this method.
With an initial focus on developing therapies for patients who cannot be treated with CFTR modulators, the partners will combine Intellia’s CRISPR-Cas9 DNA writing technology with ReCode’s selective organ targeting (SORT) LNPs to correct CFTR mutations in the lungs of patients via HDR.
A Nature Communications study published in 2023 by scientists from the University of Texas Southwestern Medical Center demonstrated proof-of-concept of this approach in mice. If their initial approach is successful, Intellia and ReCode plan to expand their program to target other CFTR mutations.
CRISPR cystic fibrosis clinical trialsThere are currently no CRISPR cystic fibrosis clinical trials. However, recent interest and investment in CF research by cell and gene therapy developers suggest that they may begin in the not-too-distant future; the announcement by Intellia and ReCode included mention of both clinical trials and commercialization.
While it can take up to a decade from proof-of-concept research to the approval of any new drug, regulatory agencies like the FDA have specific designations that can help expedite the clinical trials and review process for therapies that address significant unmet patient needs. For example, many CF patients have limited or no treatment options, meaning that any CRISPR therapy developed for the disease could potentially be given fast track, orphan drug, or priority review designations. This means we may see clinical trials for this disease begin well within the next decade.
There are currently no CRISPR cystic fibrosis clinical trials. However, recent interest and investment in CF research by cell and gene therapy developers suggest that they may begin in the not-too-distant future; the announcement by Intellia and ReCode included mention of both clinical trials and commercialization.
While it can take up to a decade from proof-of-concept research to the approval of any new drug, regulatory agencies like the FDA have specific designations that can help expedite the clinical trials and review process for therapies that address significant unmet patient needs. For example, many CF patients have limited or no treatment options, meaning that any CRISPR therapy developed for the disease could potentially be given fast track, orphan drug, or priority review designations. This means we may see clinical trials for this disease begin well within the next decade.
CRISPR Clinical Trials to Follow
New CRISPR therapies are undergoing development for genetic disorders, cancer, and infectious diseases, at all stages of the clinical workflow. We know it can be tough to keep up with all the pivotal research in this rapidly evolving field, so we've compiled a list of the top CRISPR clinical trials to watch.
The Future of CRISPR Cystic Fibrosis Research
With the advent of CRISPR technology and the recent approval of the first CRISPR therapy for a genetic disease, we have entered a new era of genomic medicine. Not only is the accurate modeling of complex diseases like CF possible, but we can also generate functional cures, offering hope to patients who previously had no treatment options.
As research continues, CRISPR will contribute to a deeper understanding of this disease, and we may see both ex vivo and in vivo approaches to CRISPR cystic fibrosis therapy enter the clinic. At Synthego, our mission is to support the development of these therapies by providing high-quality CRISPR sgRNAs for clinical use, as well as comprehensive regulatory support to help sponsors navigate the evolving regulatory landscape.
We look forward to the day that all patients with severe genetic disorders have access to life-saving treatments. Did you enjoy this blog? Explore The Bench for more articles on the applications of CRISPR in the genetic disease space, and don’t forget to subscribe for regular updates!