CRISPR technology has led to a revolution in cell and gene therapy, owing to its ability to precisely edit the genome and treat the root cause of genetic disease. But getting CRISPR gene therapy drugs to patients means progressing through an exhaustive process of development and clinical trials, which can take almost a decade.
So, are CRISPR clinical trials different from clinical trials for other drugs? And how do CRISPR gene therapy drugs get approved for patient use? Here, we address your questions about CRISPR clinical development. We’ll take you through the workflow from start to finish, explaining each stage of development and the phases of CRISPR clinical trials, FDA requirements and approval processes, and a glossary of key terms.
Discovery Research and Proof-of-Concept Studies
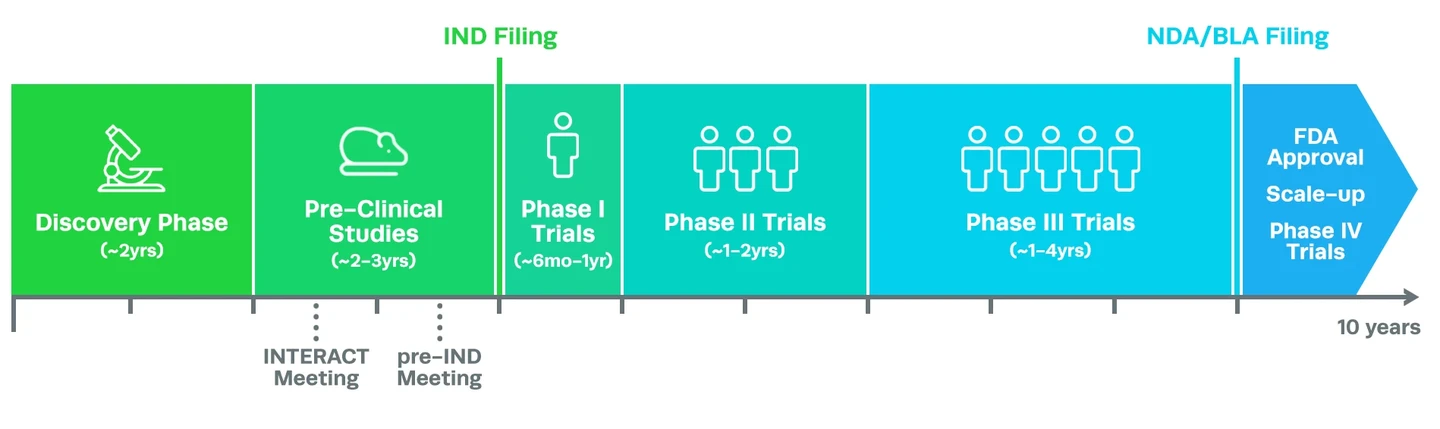
CRISPR clinical trials themselves take many years to complete, but the process of developing a cell and gene therapy product actually starts years beforehand. Proof-of-concept studies, known as discovery research, are the first major milestone in the journey. In this stage of drug discovery, researchers must pinpoint a potential treatment for a disease (target identification) and then prove that the treatment is feasible (target validation). This can take several years, especially if little is known about the disease or the genes and molecular mechanisms that cause it.
For a CRISPR gene therapy that aims to treat a genetic disorder, proof-of-concept research is typically performed in cells. The cells can either be immortalized cell lines or, better yet, primary cells taken from human patients that suffer from the disease in question. Primary cells are preferred because they more accurately reflect the biological reality of the disease compared to immortalized cells and therefore respond to potential treatments similarly.
After identifying a target, such as a genetic mutation, that causes the disease phenotype in the cells, researchers must prove not only that CRISPR gene therapy can edit the mutation at the genetic level in patient cells, but also that the editing improves the phenotype of the cells. They must also demonstrate minimal to no off-target editing or other adverse effects on the cells. If this is successful, the researchers can move on to pre-clinical research.
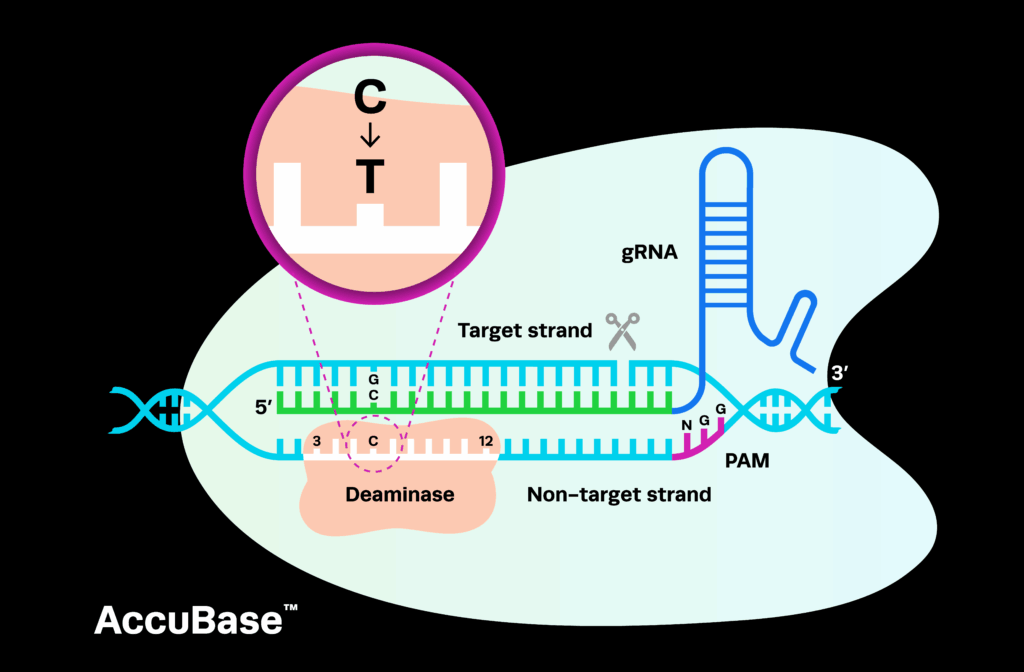
For CRISPR gene therapy and gene-edited cell therapy drugs, one of the key starting materials is the sgRNA. In discovery and proof-of-concept research, drug developers (known as ‘sponsors’) can use unregulated reagents to generate the therapy because it is only tested in cells; research use only (RUO) sgRNAs are usually sufficient for this stage of development.
Pre-Clinical Research and IND Filing
Before it’s possible to start CRISPR clinical trials, the next major stage of development for a CRISPR cell or gene therapy is to conduct rigorous pre-clinical research, typically in an animal model/s of the disease. This type of research aims to assess if the treatment works, how much is needed for a therapeutic dose, and if it is safe. This stage of development is a major challenge because cells grown in the lab respond to a potential CRISPR therapy treatment very differently compared to whole animals. As such, many potential therapies fail in this early stage.
Mammals such as mice are often the first model in which a drug will be tested, typically followed by larger animals. Some drugs also require testing in non-human primates, particularly if they affect physiological systems that are unique to primates. To ensure the treatment is effective and safe, it is essential that any animal model used in pre-clinical research accurately recapitulates the genotype, phenotype, and progression of the disease in question.
Throughout this stage of development, researchers have the opportunity to converse with the FDA about the therapy via different types of non-compulsory meetings. CRISPR cell and gene therapy drugs have complex manufacturing requirements that differ from traditional biologics, making these meetings particularly pertinent.
The first is an Initial Targeted Engagement for Regulatory Advice on CBER products (INTERACT) meeting, an informal meeting in which sponsors of CRISPR clinical trials can be advised on the chemistry, manufacturing, and controls (CMC), pharmacology and toxicology, and clinical aspects of their potential therapy by the FDA.
The second is a pre-IND meeting, the first formal meeting between the sponsor of the CRISPR clinical trial and the FDA. Pre-IND meetings are used to discuss whether the data gathered in pre-clinical development are sufficient for the FDA to approve the CRISPR gene therapy to progress to human trials when the full IND submission is made. The FDA will also advise the sponsor about current Good Manufacturing Practice (cGMP) requirements that researchers must adhere to during the development of the drug.
The data gathered during pre-clinical research is used by CRISPR therapy sponsors in their application to the FDA to begin clinical trials of the product, otherwise known as an Investigational New Drug (IND) filing. If the CRISPR-based therapy appears safe and effective in animal models, it will be given the green light to begin clinical trials in human subjects.
During pre-clinical research, the FDA often recommends that CRISPR therapy sponsors use high-quality gRNAs with some level of documentation to support their purity. To address this need, Synthego provides INDe gRNAs, which comply with the equipment, facility, and material controls in the relevant sections of 21 CFR part 58 on guidelines for Good Laboratory Practice (GLP) for nonclinical laboratory studies to be used for IND-enabling studies. When providing gRNAs for CRISPR gene therapy products, Synthego’s regulatory specialists also offer full support with the preparation of IND applications for CRISPR clinical trials.
CRISPR Clinical Trials: Phases and What They Mean
During phase I trials, the drug is usually administered to healthy subjects as well as patients with the disease in question. For more common diseases, the number of subjects can be between 20 to 80. However, for a rare genetic disease, there may be even fewer.
Clinical trials for a CRISPR therapy work much the same as any other type of drug product, progressing through phase I, II, and III trials. These different phases of CRISPR clinical trials assess key aspects of the therapy or drug - including safety, tolerability, and efficacy - in increasingly large patient populations. Each phase of a CRISPR clinical trial must be successfully completed before proceeding to the next, optimizing and refining the treatment with each step. CRISPR gene therapy and cell therapy drugs in clinical trials are required to use GMP gRNAs with extensive documentation.
The success of CRISPR clinical trials is measured according to clinical endpoints, which are pre-defined, measurable outcomes agreed upon by the sponsor and the FDA before the trials begin. Clinical endpoints vary considerably based on the disease and treatments in question. However, they can include things like increased life expectancy, increased or decreased levels of a disease-specific biomarker, or a reduction in disease symptoms.
The FDA typically has stringent criteria for allowing a new drug to proceed through the phases; 90% of drugs fail at some stage of the process. If the FDA does not receive sufficient evidence for the CRISPR therapy to progress into the next phase or there are problems with the manufacturing of the therapy, the FDA may place a clinical hold that prevents the CRISPR clinical trials from continuing until their conditions are met. If there are severe adverse events or the death of trial patients, the trial will usually be terminated unless the sponsor provides evidence that the drug was not the cause of death.
In cases where the CRISPR therapy offers a considerable benefit over what is currently available, there are limited or no available treatments, or it fulfills an urgent or unmet medical need, the FDA may agree to give the therapy a different IND designation (see glossary of terms below). These designations, such as fast track (FT) or breakthrough therapy (BT) typically help speed up the clinical trial process, sometimes by allowing two or more trial phases to run in parallel. Let’s explore the different phases of CRISPR clinical trials, what is involved, and how they are assessed.
Phase I CRISPR clinical trials: safety and dosageCRISPR clinical trials begin with phase I, where the safety of the therapy is assessed in a small group of patients over several months. Subjects are monitored very carefully to determine how the CRISPR therapy interacts with the body, what the optimum dose is, and what any acute (short-term) side effects might be. The dosage of the drug is also assessed in this phase.
CRISPR clinical trials begin with phase I, where the safety of the therapy is assessed in a small group of patients over several months. Subjects are monitored very carefully to determine how the CRISPR therapy interacts with the body, what the optimum dose is, and what any acute (short-term) side effects might be. The dosage of the drug is also assessed in this phase.
Phase II CRISPR clinical trials: efficacy and side effectsPhase II CRISPR clinical trials aim to assess the efficacy of the treatment - in other words, is the response to treatment actually beneficial? Phase II trials are performed in a larger group of up to several hundred patients. However, as we mentioned above, there may be much fewer patients in the trial for a rare genetic disease.
This phase of CRISPR clinical trials also assesses the safety of the new therapy by examining the side effects over a longer period. Phase II trials can take up to two years, and help the sponsors further refine and optimize the treatment before phase III.
Phase II CRISPR clinical trials aim to assess the efficacy of the treatment - in other words, is the response to treatment actually beneficial? Phase II trials are performed in a larger group of up to several hundred patients. However, as we mentioned above, there may be much fewer patients in the trial for a rare genetic disease.
This phase of CRISPR clinical trials also assesses the safety of the new therapy by examining the side effects over a longer period. Phase II trials can take up to two years, and help the sponsors further refine and optimize the treatment before phase III.
Phase III CRISPR clinical trials: efficacy and monitoring for adverse eventsPhase III CRISPR clinical trials are the final steps when researchers are developing a new CRISPR therapy, but they also take the longest of any trial phase - up to four years in some cases. This phase is also performed in the largest group of patients, usually between 300 to 3,000. In this phase, the efficacy of the therapy is assessed and compared to other current treatment options.
For the CRISPR gene therapy to be approved, it must be demonstrated to provide significant benefits over what is currently available. For many rare genetic diseases, there may be no other available treatments, making comparison in CRISPR clinical trials impossible. The CRISPR therapy must also be low-risk, with few or manageable side effects. The occurrence of side effects and adverse events are monitored long-term, over several years.
Phase III CRISPR clinical trials are the final steps when researchers are developing a new CRISPR therapy, but they also take the longest of any trial phase - up to four years in some cases. This phase is also performed in the largest group of patients, usually between 300 to 3,000. In this phase, the efficacy of the therapy is assessed and compared to other current treatment options.
For the CRISPR gene therapy to be approved, it must be demonstrated to provide significant benefits over what is currently available. For many rare genetic diseases, there may be no other available treatments, making comparison in CRISPR clinical trials impossible. The CRISPR therapy must also be low-risk, with few or manageable side effects. The occurrence of side effects and adverse events are monitored long-term, over several years.
Gaining FDA approval and phase IV CRISPR clinical trialsAt this point, more than eight years may have passed since the beginning of the clinical development process. After phase III CRISPR trials, the complete clinical trial data are presented to the FDA for a final assessment in a New Drug Application (NDA). The benefits of the CRISPR therapy (based on the agreed endpoints or surrogate endpoints) are weighed against current treatment options, as well as the known acute and chronic side effects of the drug before the FDA either rejects the therapy or approves it for medical use.
Counter-intuitively, phase IV clinical trials commonly take place after a therapy has received FDA approval. If the FDA approves the CRISPR therapy and it is produced and marketed to the public, phase IV is the post-clinical safety monitoring of the drug in the wider patient population.
At this point, more than eight years may have passed since the beginning of the clinical development process. After phase III CRISPR trials, the complete clinical trial data are presented to the FDA for a final assessment in a New Drug Application (NDA). The benefits of the CRISPR therapy (based on the agreed endpoints or surrogate endpoints) are weighed against current treatment options, as well as the known acute and chronic side effects of the drug before the FDA either rejects the therapy or approves it for medical use.
Counter-intuitively, phase IV clinical trials commonly take place after a therapy has received FDA approval. If the FDA approves the CRISPR therapy and it is produced and marketed to the public, phase IV is the post-clinical safety monitoring of the drug in the wider patient population.
Commercial Manufacturing of CRISPR Therapies

While it may seem that gaining FDA approval is the biggest challenge in the CRISPR clinical trials process, in reality, the commercial manufacturing of the CRISPR therapy can be just as difficult. Creating a product that meets demand requires efficient scaling-up while maintaining stringent quality control measures, and this is where many sponsors run into issues.
For example, when a gene-edited cell therapy is being developed by a sponsor, the FDA may specify that the commercial therapy contain a certain threshold of viable cells. If the final product does not meet this threshold, the sponsor will not be able to market the product. In some cases, the therapy can still be provided to patients for compassionate use, but the sponsor can’t profit from it.
CRISPR Cell and Gene Therapies: Key Challenges in Clinical Development
Researchers face many hurdles when developing CRISPR therapy products, including the procurement of true GMP reagents, unclear regulatory guidelines, lack of consistency during development, and shortages of qualified experts. Read more about these issues in CRISPR therapy development and strategies to overcome them.

The Future of CRISPR Therapies
In December 2023, we saw the first US approval of a CRISPR therapy developed by CRISPR Therapeutics and Vertex Pharmaceuticals, also approved in Europe. The CRISPR gene therapy drug called Casgevy (exa-cel) is an ex vivo cell therapy for the treatment of sickle cell disease and beta-thalassemia. Many more CRISPR gene therapy and cell therapy drugs are currently in clinical trials, making the next few years a particularly exciting time for CRISPR clinical trials as we see more results trickle in. If you’ve been keeping up with the broader CRISPR field, you may have also noticed an increase in discovery and pre-clinical research using CRISPR derivative technologies, such as base editing, prime editing, and PASTE. These spin-offs of CRISPR still use gRNAs to direct editing, but do not induce DSBs. We’ll likely see more of these alternatives enter the clinic in the coming years.
CRISPR Clinical Trials to Follow
New CRISPR therapies are undergoing development for genetic disorders, cancer, and infectious diseases, at all stages of the clinical workflow. We know it can be tough to keep up with all the pivotal research in this rapidly evolving field, so we've compiled a list of the top CRISPR clinical trials to watch.
As a trusted leader in genome editing technology, Synthego’s mission is to help accelerate the process of clinical development for CRISPR gene therapy products, reducing costs and making these therapies as safe as possible. We partner with you to provide support and documentation at every step of the clinical development process, from pre-IND through to phase IV CRISPR clinical trials.
To kickstart your CRISPR therapy development process, get in touch with our CRISPR experts.
Glossary of Drug Designations and Other Key Terms
BLA: Biologics License Applications. BLAs request permission for biologics to be introduced and delivered into interstate commerce, and includes similar documentation to an NDA.
CBER: Center for Biologics Evaluation and Research. This branch of the FDA deals with biologics and related therapeutics, ensuring their safety and efficacy.
IND: Investigational New Drug application. This application is submitted to the FDA and includes preclinical data to support the therapy entering clinical trials in human subjects.
cGMP: Refers to Current Good Manufacturing Practice, the primary regulatory standard for ensuring the safety, quality, strength, identity, and purity of pharmaceutical products, including cell and gene therapies. All therapies in clinical trials must adhere to cGMP requirements.
Fast track status: An FDA designation for therapies that treat serious conditions and/or fulfill unmet medical needs. For example, if there are no currently available treatments for the condition, or if the therapy will have a serious impact on the day-to-day functioning or survival of patients, it may be given this designation. Fast track designation speeds up the development and review of the therapy to get the new drug to patients sooner.
Breakthrough therapy (BT): This designation speeds up the development and review process for a therapy if early evidence from clinical trials indicates a clear and substantial benefit compared to currently available therapies. This can include if the drug has significantly reduced side effects but has similar efficacy to currently available treatments. Drugs designated as breakthrough therapies can be approved based on surrogate or intermediate endpoints.
Accelerated approval: A regulation that allows for a new therapy to be approved faster by the FDA based on surrogate or intermediate endpoints. Accelerated approval can be granted if the therapy treats a serious condition or fulfills an unmet medical need.
Priority review: A priority review designation means that the FDA aims to make a decision about the therapy in question within six months of the final application being submitted for review. This designation can be given to drugs that can demonstrate a significant improvement in safety compared to currently available treatments. Drugs that are not given this designation are subject to standard review, which takes 10 months.
Rolling review: Rather than waiting for the complete clinical trial data to submit an NDA or BLA application, rolling review is a process in which smaller sections of these applications are submitted to the FDA as they are completed, helping to speed up the review process further. Drugs which are granted fast track designation are also eligible for rolling review.
Surrogate and intermediate endpoints: Surrogate and intermediate endpoints are used in place of normal clinical endpoints when a drug is subject to certain designations such as accelerated approval or breakthrough therapy. Surrogate endpoints are markers that predict clinical benefit, rather than measurements of proven clinical benefit. They can include markers such as radiographic imaging or laboratory measurements. Similarly, intermediate endpoints measure the effects of the therapy that are reasonable indicators of the clinical benefit.
RMAT: Regenerative Medicine Advanced Therapy designation. This designation is particularly relevant to CRISPR clinical trials because it applies specifically to cell therapies, engineered therapeutic tissues, and other human cell and tissue products.
Orphan drug designation/rare disease designation: This designation can be given to therapies that diagnose, prevent, or treat rare diseases, usually when there are limited or no other available alternatives. If granted an orphan drug designation, the sponsors of the therapy qualify for major drug development incentives, such as tax credits for the clinical trials process, fee exemptions, and market exclusivity for up to seven years after the drug is approved.
Adverse events: Adverse events are defined by the FDA as any unfavorable or unintended medical occurrence associated with the use of a drug, even if it is not considered to be caused by the drug. This can include abnormal laboratory findings, symptoms, or other factors. Serious adverse events are more severe occurrences, such as disability, permanent damage, birth defects, hospitalization, death, or life-threatening situations as a result of the drug.
NDA: New Drug Application. This is the documentation of all the data from preclinical research and clinical trials that are presented to the FDA for final approval. It helps the FDA make key decisions about the safety and efficacy of the therapy, its potential risks, whether the product labeling and information are appropriate, and whether the manufacturing and purity of the drug meet current standards.
Clinical hold: An order issued by the FDA to IND sponsors that will either delay or suspend any clinical trials for that therapy. A clinical hold can be issued because of unacceptable levels of risk to patients, lack of qualification of the sponsor to conduct the trials properly, failure to comply with FDA standards such as cGMP requirements, misleading or incomplete product information, risk of reproductive toxicity as the result of the therapy, or deficiencies in the design of the trial.