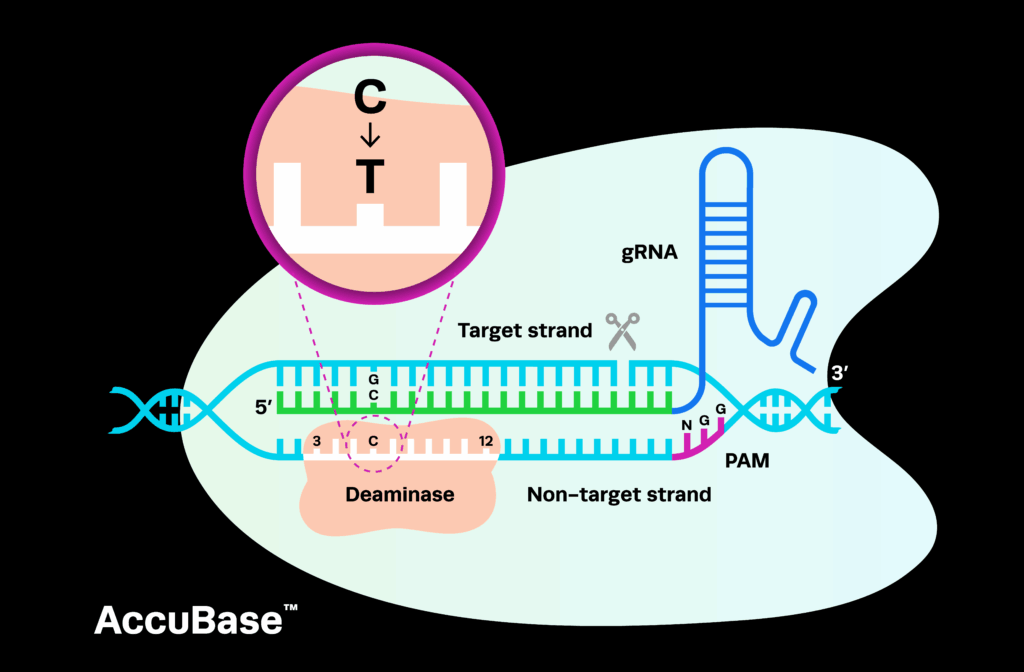
CRISPR cell and gene therapy drugs are revolutionizing medicine, and the clinical landscape has shifted considerably in the last few years. However, staying up-to-date with all the developments in the regulatory space and understanding how these developments will affect cell and gene therapy products can be a challenge.
Here, we cover key regulatory developments in the cell and gene therapy space, particularly FDA initiatives concerning these novel biologics, the publication of FDA guidance documents that may help accelerate their development, and other regulatory trends including the release of an internationally-recognized lexicon of gene editing terms.
Growth and Challenges in Cell & Gene Therapy Development
In December of 2023, the FDA approved Casgevy for the treatment of sickle cell disease (SCD). The first CRISPR-based therapy to be approved for widespread use, Cagevy (also known as exa-cel) is an autologous gene-edited cell therapy that upregulates the production of fetal hemoglobin. This historic approval set a precedent for the development and approval of even more CRISPR-based medicines, with investigational new drug (IND) filings expected to increase in the coming years. It also allowed regulatory bodies like the FDA to develop a framework for how future CRISPR therapies can be regulated and approved.
Despite the success of Casgevy, there remain several key challenges in the CRISPR cell and gene therapy field. Bottlenecks in the manufacturing process, communication issues between the FDA and sponsors, and shortages of qualified specialist staff have caused many therapies to falter at critical stages of the clinical development pipeline. In addition to the challenges faced by researchers and companies, the FDA is known to be chronically understaffed, making it difficult to get therapies through the review process in a timely manner.
Another key problem in the industry is the financial feasibility of generating small-batch CGT products for ‘ultra-rare’ diseases with limited patient populations. Many companies are unable to develop these niche therapies because of the associated costs and limited financial return.

A survey conducted by Synthego revealed that dealing with confusing regulatory guidelines is one of the top obstacles faced by researchers developing CGT products, closely followed by the accessibility of good manufacturing practice (GMP)-grade reagents and issues with clinical trial site management.
Understanding FDA Guidances on Cell & Gene Therapy
The FDA has released several key guidance documents concerning the development of CRISPR cell and gene therapies in the last several years. However, FDA guidances can be confusing, even if you’re familiar with the cell and gene therapy space. Here, we will explore these FDA guidance documents and why they're important for developers of CRISPR medicines.
FDA Releases Final Guidance on Early-Phase Cell and Gene Therapy TrialsIn November 2022, the Center for Biologics Evaluation and Research (CBER) released a final FDA guidance for the industry that provides important recommendations for developers of cell and gene therapy products (known as ‘sponsors’). Let’s take an in-depth look at why this guidance was released, what recommendations are in the guidance, and what this means for cell and gene therapy development, particularly CRISPR gene therapy drugs.
In November 2022, the Center for Biologics Evaluation and Research (CBER) released a final FDA guidance for the industry that provides important recommendations for developers of cell and gene therapy products (known as ‘sponsors’). Let’s take an in-depth look at why this guidance was released, what recommendations are in the guidance, and what this means for cell and gene therapy development, particularly CRISPR gene therapy drugs.
Reason behind the recent FDA cell and gene therapy guidanceTitled ‘Studying Multiple Versions of a Cellular or Gene Therapy Product in an Early-Phase Clinical Trial’, this particular guidance came in response to interest from sponsors in umbrella trials, where multiple versions of a drug or therapy are tested in the same preliminary trial. The guidance refers to a key example of chimeric antigen receptor (CAR) T cell therapy, a form of cellular immunotherapy used to treat cancer, where there may be slightly different versions of the therapy based on the specific CAR protein domain that may increase the anti-tumor activity of the therapy. Under an early-phase umbrella trial, each version of the CAR-T cell therapy can be assessed in parallel rather than in separate trials with a different control group for each.
Titled ‘Studying Multiple Versions of a Cellular or Gene Therapy Product in an Early-Phase Clinical Trial’, this particular guidance came in response to interest from sponsors in umbrella trials, where multiple versions of a drug or therapy are tested in the same preliminary trial. The guidance refers to a key example of chimeric antigen receptor (CAR) T cell therapy, a form of cellular immunotherapy used to treat cancer, where there may be slightly different versions of the therapy based on the specific CAR protein domain that may increase the anti-tumor activity of the therapy. Under an early-phase umbrella trial, each version of the CAR-T cell therapy can be assessed in parallel rather than in separate trials with a different control group for each.
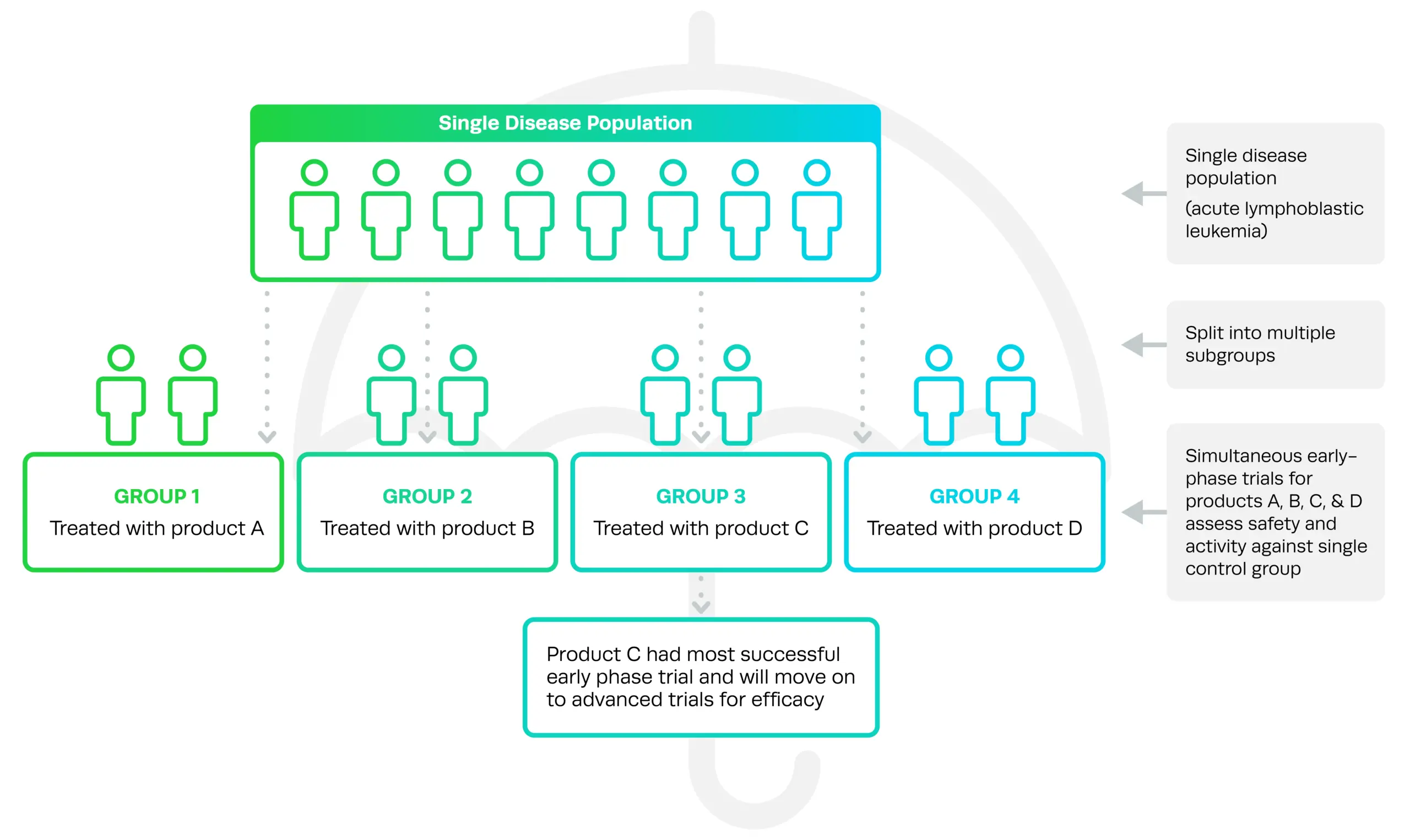
Common in the cancer therapy space for many years—and more recently in COVID-19 studies—umbrella trials allow sponsors to directly compare different versions of a drug and choose the best to push forward into advanced trials.
Draft versions of FDA guidances are typically released early in order to give sponsors, advocacy groups, and other industry members a chance to respond and ask questions before the final version is published. After releasing a draft version of these recommendations in September of 2021, the FDA has addressed comments and concerns from industry representatives in the final guidance. The guidance is specific to cell and gene therapies, not other products such as small-molecule drugs.
What’s in the FDA cell and gene therapy guidance, and why is it important?Umbrella trials work on the basis of a ‘master protocol’, which aims to evaluate the different versions of a therapy or drug simultaneously to determine which is the safest or most effective. These different versions will be treated as separate drug products, but can still be assessed simultaneously by a master protocol in a single trial.
The new guidance clarifies several aspects of umbrella trial design for cell and gene therapy development. This includes how Investigational New Drug (IND) applications for umbrella trials should be structured and organized in order to avoid unnecessarily duplicating documentation.
Under the umbrella trial structure, there is one Primary IND A, which contains the master protocol information that all versions of the therapy will be tested under, and CMC and pharmacology/toxicity (P/T) information for one version of the therapy, product A. The secondary INDs contain CMC and P/T information on the other versions of the therapy (B, C, D, and so on), and can be cross-referenced to the Primary IND A for information about the master protocol.
Umbrella trials work on the basis of a ‘master protocol’, which aims to evaluate the different versions of a therapy or drug simultaneously to determine which is the safest or most effective. These different versions will be treated as separate drug products, but can still be assessed simultaneously by a master protocol in a single trial.
The new guidance clarifies several aspects of umbrella trial design for cell and gene therapy development. This includes how Investigational New Drug (IND) applications for umbrella trials should be structured and organized in order to avoid unnecessarily duplicating documentation.
Under the umbrella trial structure, there is one Primary IND A, which contains the master protocol information that all versions of the therapy will be tested under, and CMC and pharmacology/toxicity (P/T) information for one version of the therapy, product A. The secondary INDs contain CMC and P/T information on the other versions of the therapy (B, C, D, and so on), and can be cross-referenced to the Primary IND A for information about the master protocol.
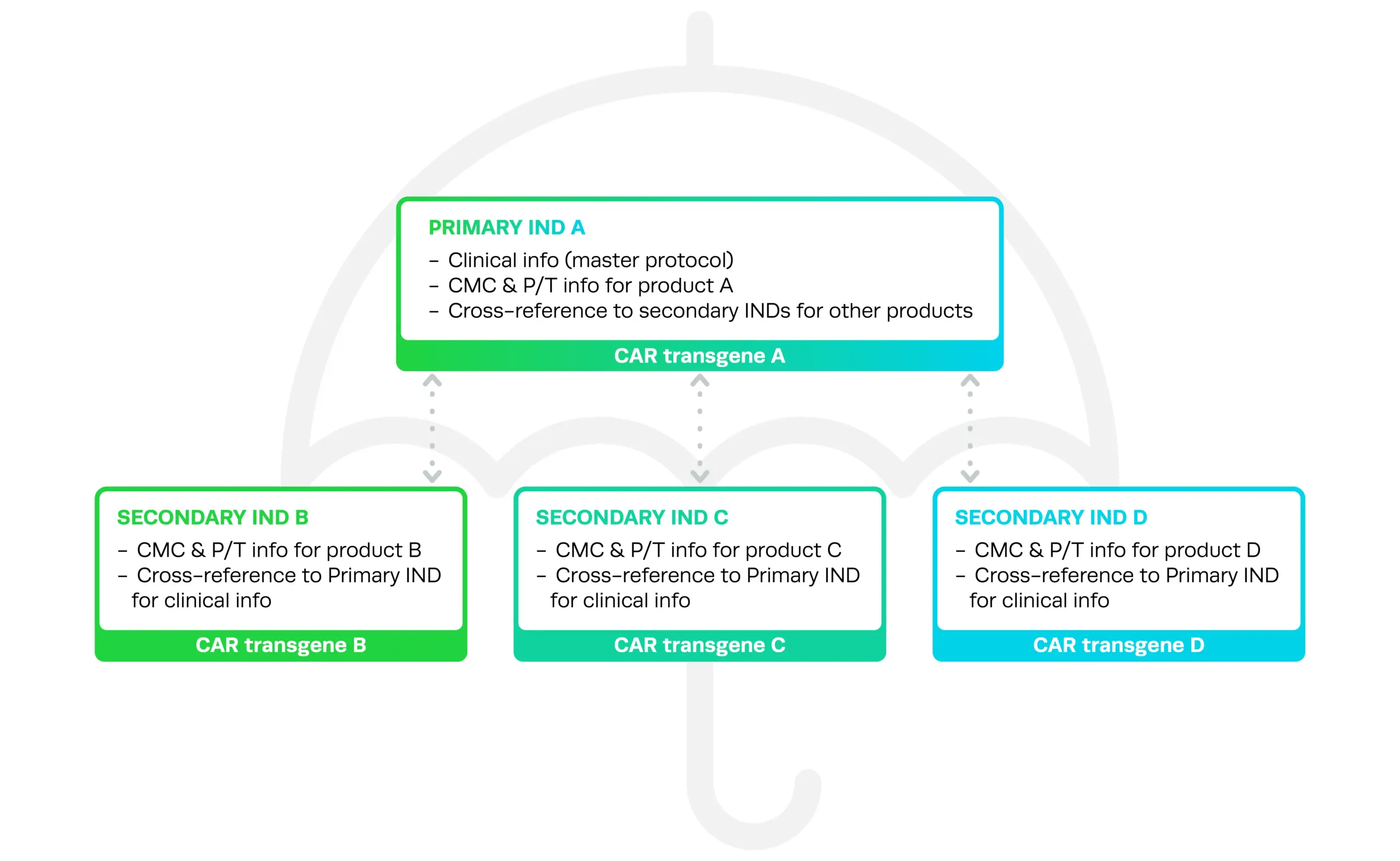
The guidance also explains how to add and update study arms or other information during development, the consequences for the entire umbrella trial if one version is subject to a clinical hold, and how to report on the safety of umbrella trials effectively.
Importantly, the guidance also clarifies what constitutes different versions of a cell or gene therapy product and what does not, so that sponsors can more efficiently prepare INDs and avoid duplication where it isn’t necessary. For example, changing from adherent to suspension cell culture during product development is not considered a different version of the therapy and does not require separate INDs.
In contrast, in vivo CRISPR gene therapy drugs are commonly delivered via adeno-associated viral vectors (AAVs). A sponsor may be investigating two different versions of the AAV where a viral capsid protein has been mutated, to see which version is more efficient for delivery or has the lowest immune response in patients. According to the guidance, these will be treated as different versions of the drug, even if the CRISPR reagents contained within the two types of AAV are exactly the same. This means they will require separate INDs, but can still be assessed in the same umbrella trial.
How will the FDA cell and gene therapy guidance affect CRISPR therapy development?The appeal of running an umbrella trial is primarily that it will accelerate the drug development process. Running trials is expensive and time-consuming, particularly for advanced therapies such as CRISPR gene therapy products and gene-edited cell therapies; testing different versions of a drug simultaneously can maximize efficiency in terms of both time and cost.
Another key advantage is that based on a master protocol, different versions of a CRISPR gene therapy could be tested using the same control group for comparison, while previously, each would be compared to its own separate control group. This means that early-phase studies will require the recruitment of fewer subjects; in the rare disease space, recruiting enough participants in a study is often a challenge.
FDA’s support for umbrella trials in the rare disease space is encouraging; it’s certainly evident that they are listening to feedback from cell and gene therapy sponsors and helping streamline the process of clinical development. This is welcome news, considering the recent impetus for a restructuring of outdated clinical frameworks for novel CRISPR gene therapies, a cause championed by leading scientists like Professor Fyodor Urnov of the Innovative Genomics Institute.
Importantly, this guidance could eventually result in an uptick in the number of approved CRISPR gene therapies available to patients with cancer and genetic disorders. Using umbrella trials to accelerate the clinical development pipeline for complex cell and gene therapy products means that sponsors can get these drugs to patients faster and hopefully with a slightly lower price tag.
The second draft guidance is called ‘Considerations for the Development of Chimeric Antigen Receptor (CAR) T Cell Products’. While the title refers specifically to CAR-T cell therapies, which are used to treat cancer, the FDA remarked that these guidelines are also more broadly applicable to other gene-edited cell therapies, such as natural killer (NK) and T cell receptor (TCR) cell therapies. The document includes recommendations on safety, manufacturing, clinical study design, and analytical comparability of CAR-T products.
The appeal of running an umbrella trial is primarily that it will accelerate the drug development process. Running trials is expensive and time-consuming, particularly for advanced therapies such as CRISPR gene therapy products and gene-edited cell therapies; testing different versions of a drug simultaneously can maximize efficiency in terms of both time and cost.
Another key advantage is that based on a master protocol, different versions of a CRISPR gene therapy could be tested using the same control group for comparison, while previously, each would be compared to its own separate control group. This means that early-phase studies will require the recruitment of fewer subjects; in the rare disease space, recruiting enough participants in a study is often a challenge.
FDA’s support for umbrella trials in the rare disease space is encouraging; it’s certainly evident that they are listening to feedback from cell and gene therapy sponsors and helping streamline the process of clinical development. This is welcome news, considering the recent impetus for a restructuring of outdated clinical frameworks for novel CRISPR gene therapies, a cause championed by leading scientists like Professor Fyodor Urnov of the Innovative Genomics Institute.
Importantly, this guidance could eventually result in an uptick in the number of approved CRISPR gene therapies available to patients with cancer and genetic disorders. Using umbrella trials to accelerate the clinical development pipeline for complex cell and gene therapy products means that sponsors can get these drugs to patients faster and hopefully with a slightly lower price tag.
The second draft guidance is called ‘Considerations for the Development of Chimeric Antigen Receptor (CAR) T Cell Products’. While the title refers specifically to CAR-T cell therapies, which are used to treat cancer, the FDA remarked that these guidelines are also more broadly applicable to other gene-edited cell therapies, such as natural killer (NK) and T cell receptor (TCR) cell therapies. The document includes recommendations on safety, manufacturing, clinical study design, and analytical comparability of CAR-T products.
New draft guidance for gene therapies and CAR-T cell therapiesIn 2022, the FDA also released two new draft guidance documents to help sponsors who are developing CGT products. The first is titled ‘Human Gene Therapy Products Incorporating Human Genome Editing’, and provides recommendations on the information that should be included in IND filings for these therapies, including study design, safety, and manufacturing. This guidance has since been finalized.
The second draft guidance is called ‘Considerations for the Development of Chimeric Antigen Receptor (CAR) T Cell Products’. While the title refers specifically to CAR-T cell therapies, which are used to treat cancer, the FDA remarked that these guidelines are also more broadly applicable to other gene-edited cell therapies, such as natural killer (NK) and T cell receptor (TCR) cell therapies. The document includes recommendations on safety, manufacturing, clinical study design, and analytical comparability of CAR-T products.
In 2022, the FDA also released two new draft guidance documents to help sponsors who are developing CGT products. The first is titled ‘Human Gene Therapy Products Incorporating Human Genome Editing’, and provides recommendations on the information that should be included in IND filings for these therapies, including study design, safety, and manufacturing. This guidance has since been finalized.
The second draft guidance is called ‘Considerations for the Development of Chimeric Antigen Receptor (CAR) T Cell Products’. While the title refers specifically to CAR-T cell therapies, which are used to treat cancer, the FDA remarked that these guidelines are also more broadly applicable to other gene-edited cell therapies, such as natural killer (NK) and T cell receptor (TCR) cell therapies. The document includes recommendations on safety, manufacturing, clinical study design, and analytical comparability of CAR-T products.
FDA Initiatives in Cell & Gene Therapy
The release of FDA guidance documents isn’t the only recent development in the cell and gene therapy space – the FDA have had major office revamps, introduced several initiatives focused on accelerating the development of cell and gene therapies, and tried to improve communications through a range of public and private meetings with CRISPR therapy sponsors.
FDA’s OTAT becomes the Office of Therapeutic Products (OTP)The FDA’s Office of Tissues and Advanced Therapies (OTAT), which was responsible for regulating cell and gene therapy products, has had a major revamp in the last couple of years, establishing the new Office of Therapeutic Products (OTP). This ‘super office’ was designed to have increased review capabilities and more expertise - in cell and gene therapy products in particular. The OTP has six sub-offices, covering gene therapy CMC, cellular therapy and human tissue CMC, plasma protein therapeutics CMC, clinical evaluation, pharmacology/toxicology, and review management and regulatory review.
To deal with the surge in cell and gene therapy applications, the FDA advertised more than 100 new positions, and as of early 2024, the OTP was 75-80% staffed. The regulatory body has stated that it also aims to provide opportunities for career advancement so that it can better recruit and retain qualified staff, which has been problematic due to competitive industry salaries.
An increase in qualified staff at the FDA should hopefully lead to accelerated review times for complex biologics like CRISPR gene therapy drugs, as well as more valuable feedback for the sponsors of these therapies. The OTP has further stated its commitment to supporting the development of cell and gene therapies, and forging collaborations and partnerships with industry sponsors. The FDA also developed a communications pilot program for rare disease therapies called Support for clinical Trials Advancing Rare disease Therapeutics (START). With a goal of accelerating the pace of development for rare diseases in which there is significant unmet medical need, START allows sponsors to ask reviewers for advice on product-specific development issues.
The FDA’s Office of Tissues and Advanced Therapies (OTAT), which was responsible for regulating cell and gene therapy products, has had a major revamp in the last couple of years, establishing the new Office of Therapeutic Products (OTP). This ‘super office’ was designed to have increased review capabilities and more expertise - in cell and gene therapy products in particular. The OTP has six sub-offices, covering gene therapy CMC, cellular therapy and human tissue CMC, plasma protein therapeutics CMC, clinical evaluation, pharmacology/toxicology, and review management and regulatory review.
To deal with the surge in cell and gene therapy applications, the FDA advertised more than 100 new positions, and as of early 2024, the OTP was 75-80% staffed. The regulatory body has stated that it also aims to provide opportunities for career advancement so that it can better recruit and retain qualified staff, which has been problematic due to competitive industry salaries.
An increase in qualified staff at the FDA should hopefully lead to accelerated review times for complex biologics like CRISPR gene therapy drugs, as well as more valuable feedback for the sponsors of these therapies. The OTP has further stated its commitment to supporting the development of cell and gene therapies, and forging collaborations and partnerships with industry sponsors. The FDA also developed a communications pilot program for rare disease therapies called Support for clinical Trials Advancing Rare disease Therapeutics (START). With a goal of accelerating the pace of development for rare diseases in which there is significant unmet medical need, START allows sponsors to ask reviewers for advice on product-specific development issues.
Improving communications and meeting opportunities with FDACommunication issues between sponsors and the FDA has been an ongoing issue in the development of cell and gene therapies. For many researchers and sponsors, progressing from pre-clinical research into clinical trials can be an extremely challenging process, not least because of unclear regulatory guidelines. Fortunately, the FDA is now prioritizing more effective communications with sponsors of CGT products, encouraging a growth trajectory in the coming years.
This included a plan to provide increased guidance for sponsors by facilitating a variety of workshops and webinars. Increased consistency in the review process and timeliness of reviews have also been touted as key priorities, along with broader public messaging surrounding these therapies. Through these initiatives and further engagement with the industry, the FDA hopes to enable the development and, most importantly, success, of more complex CGT products, bringing them to patients sooner.
A key initiative in this space is the INitial Targeted Engagement for Regulatory Advice on CBER producTs (INTERACT) program. This program was essentially an attempt to address the growing pains experienced by the CGT industry within current clinical frameworks and involves informal meetings between CBER/OTP staff and researchers/sponsors who are at the pre-IND stage of development.
There are now a variety of opportunities for sponsors to meet with regulators throughout the process of clinical development, both in formal and informal settings. This includes INTERACT and pre-IND meetings, meetings at the end of each phase of clinical trials, and pre-BLA meetings.
Communication issues between sponsors and the FDA has been an ongoing issue in the development of cell and gene therapies. For many researchers and sponsors, progressing from pre-clinical research into clinical trials can be an extremely challenging process, not least because of unclear regulatory guidelines. Fortunately, the FDA is now prioritizing more effective communications with sponsors of CGT products, encouraging a growth trajectory in the coming years.
This included a plan to provide increased guidance for sponsors by facilitating a variety of workshops and webinars. Increased consistency in the review process and timeliness of reviews have also been touted as key priorities, along with broader public messaging surrounding these therapies. Through these initiatives and further engagement with the industry, the FDA hopes to enable the development and, most importantly, success, of more complex CGT products, bringing them to patients sooner.
A key initiative in this space is the INitial Targeted Engagement for Regulatory Advice on CBER producTs (INTERACT) program. This program was essentially an attempt to address the growing pains experienced by the CGT industry within current clinical frameworks and involves informal meetings between CBER/OTP staff and researchers/sponsors who are at the pre-IND stage of development.
There are now a variety of opportunities for sponsors to meet with regulators throughout the process of clinical development, both in formal and informal settings. This includes INTERACT and pre-IND meetings, meetings at the end of each phase of clinical trials, and pre-BLA meetings.
Town hall meetings on clinical development of cell and gene therapies Before it became the OTP, the OTAT hosted two key town hall meetings concerning Chemistry, Manufacturing, and Control (CMC) requirements, one for cell therapies and one for gene therapies. Beyond the various meetings it has with individual sponsors about specific therapies, these public meetings provide an informal space for sponsors to ask a variety of questions.
The CMC module forms a key component of the Common Technical Document (CTD), which is required by the FDA for all IND and Biologics License Application (BLA) submissions. Developers of cell and gene therapy drugs - particularly CRISPR therapy products - often find regulatory paperwork like CMC modules a major headache. During the meetings, OTAT representatives answered various questions submitted by attendees about CMC requirements for cell and gene therapies. The sheer number of questions submitted was evidence of how many sponsors struggle with regulatory requirements and guidelines.
Topics covered during the meetings included the requirements for Good Manufacturing Practice (GMP)-grade reagents, experimental design for clinical trials, quality control testing and risk assessment, use of stem cells and induced pluripotent stem cells (iPSCs), sourcing of allogeneic (donor) cells for therapeutic use, commercial specifications for gene therapies, and viral delivery of cell and gene therapy products.
In 2023, OTAT and CBER hosted another public town hall meeting in which they discussed the clinical development of gene therapy products for rare diseases. Representatives from OTAT and CBER answered various questions from sponsors of gene therapy products relating to the FDA guidance on the subject released in 2020, titled Human Gene Therapy for Rare Diseases.
A short time later, CBER’s then-director Peter Marks announced that the department will soon launch a new Operation Warp Speed pilot program for rare diseases, addressing areas of unmet need. While the original Operation Warp Speed was a government-industry partnership for the rapid development of COVID-19 vaccines, this iteration aims to accelerate the development of novel biologics, such as CRISPR gene therapy drugs, for genetic disorders.
There have since been several more virtual town hall meetings held by the OTP, covering related topics such as best practices for regulatory interactions, cell therapy CMC readiness for late-stage INDs, nonclinical assessment of cell and gene therapy products, and CMC readiness for gene therapy BLAs.
Before it became the OTP, the OTAT hosted two key town hall meetings concerning Chemistry, Manufacturing, and Control (CMC) requirements, one for cell therapies and one for gene therapies. Beyond the various meetings it has with individual sponsors about specific therapies, these public meetings provide an informal space for sponsors to ask a variety of questions.
The CMC module forms a key component of the Common Technical Document (CTD), which is required by the FDA for all IND and Biologics License Application (BLA) submissions. Developers of cell and gene therapy drugs - particularly CRISPR therapy products - often find regulatory paperwork like CMC modules a major headache. During the meetings, OTAT representatives answered various questions submitted by attendees about CMC requirements for cell and gene therapies. The sheer number of questions submitted was evidence of how many sponsors struggle with regulatory requirements and guidelines.
Topics covered during the meetings included the requirements for Good Manufacturing Practice (GMP)-grade reagents, experimental design for clinical trials, quality control testing and risk assessment, use of stem cells and induced pluripotent stem cells (iPSCs), sourcing of allogeneic (donor) cells for therapeutic use, commercial specifications for gene therapies, and viral delivery of cell and gene therapy products.
In 2023, OTAT and CBER hosted another public town hall meeting in which they discussed the clinical development of gene therapy products for rare diseases. Representatives from OTAT and CBER answered various questions from sponsors of gene therapy products relating to the FDA guidance on the subject released in 2020, titled Human Gene Therapy for Rare Diseases.
A short time later, CBER’s then-director Peter Marks announced that the department will soon launch a new Operation Warp Speed pilot program for rare diseases, addressing areas of unmet need. While the original Operation Warp Speed was a government-industry partnership for the rapid development of COVID-19 vaccines, this iteration aims to accelerate the development of novel biologics, such as CRISPR gene therapy drugs, for genetic disorders.
There have since been several more virtual town hall meetings held by the OTP, covering related topics such as best practices for regulatory interactions, cell therapy CMC readiness for late-stage INDs, nonclinical assessment of cell and gene therapy products, and CMC readiness for gene therapy BLAs.
The Bespoke Gene Therapy Consortium (BGTC) and a rare disease pilot program The FDA announced another important initiative as part of its NIH Accelerating Medicines Partnership Program. A public-private partnership between the FDA, NIH, industry, and nonprofit partners, the Bespoke Gene Therapy Consortium (BGTC) aims to overcome major obstacles and streamline the development process for small-batch gene therapies.
While each ultra-rare genetic disorder may have a very small patient population, when these conditions are grouped together, they make up a significant number. The FDA has indicated that financial incentives are also required in order to encourage companies to create therapies that are not ‘commercially viable’. The BGTC was designed to act as a ‘cookbook’ for these therapies, providing information on basic and clinical research, manufacturing, and production, and regulatory requirements, specifically concerning the use of adeno-associated viral vectors (AAVs) for delivery.
The idea of generating gene therapies for very small patient populations has been championed by several researchers in the CRISPR field, including Professor Fyodor Urnov of UC Berkeley’s Innovative Genomics Institute. Urnov has also suggested that an entirely new clinical framework is required for edited CGTs in order to get treatments to patients faster.
The FDA announced another important initiative as part of its NIH Accelerating Medicines Partnership Program. A public-private partnership between the FDA, NIH, industry, and nonprofit partners, the Bespoke Gene Therapy Consortium (BGTC) aims to overcome major obstacles and streamline the development process for small-batch gene therapies.
While each ultra-rare genetic disorder may have a very small patient population, when these conditions are grouped together, they make up a significant number. The FDA has indicated that financial incentives are also required in order to encourage companies to create therapies that are not ‘commercially viable’. The BGTC was designed to act as a ‘cookbook’ for these therapies, providing information on basic and clinical research, manufacturing, and production, and regulatory requirements, specifically concerning the use of adeno-associated viral vectors (AAVs) for delivery.
The idea of generating gene therapies for very small patient populations has been championed by several researchers in the CRISPR field, including Professor Fyodor Urnov of UC Berkeley’s Innovative Genomics Institute. Urnov has also suggested that an entirely new clinical framework is required for edited CGTs in order to get treatments to patients faster.
Other Regulatory Developments and Trends
Outside of FDA initiatives and changes, there have been plenty of other interesting and useful occurrences in the regulatory space when it comes to CRISPR therapies.
NIST Standardizes Genome Editing TermsThe end of 2022 saw a key development in the harmonization of terms in the genome editing space. The Genome Editing Consortium, established by the US National Institute of Standards and Technology (NIST), published an official lexicon of genome editing terminology, including 42 key terms. Supported by the International Standards Organization (ISO), which develops and publishes international standards for technology and manufacturing, the vocabulary was also verified by experts from various countries. The harmonization of terms aims to minimize confusion surrounding genome editing technology like CRISPR gene therapy products, both within the field itself and in the broader community, perhaps contributing to the broader acceptance of CRISPR cell and gene therapy drugs. Listen to Dr. Samantha Maragh, Leader of the Genome Editing Program, discuss the Genome Editing Consortium’s work, including the development of the lexicon, in her talk at World CRISPR Day 2022 (3:58:48 time point).
The end of 2022 saw a key development in the harmonization of terms in the genome editing space. The Genome Editing Consortium, established by the US National Institute of Standards and Technology (NIST), published an official lexicon of genome editing terminology, including 42 key terms. Supported by the International Standards Organization (ISO), which develops and publishes international standards for technology and manufacturing, the vocabulary was also verified by experts from various countries. The harmonization of terms aims to minimize confusion surrounding genome editing technology like CRISPR gene therapy products, both within the field itself and in the broader community, perhaps contributing to the broader acceptance of CRISPR cell and gene therapy drugs. Listen to Dr. Samantha Maragh, Leader of the Genome Editing Program, discuss the Genome Editing Consortium’s work, including the development of the lexicon, in her talk at World CRISPR Day 2022 (3:58:48 time point).
Regulatory harmonization and agreement on surrogate endpoints
A major topic of discussion in the cell and gene therapy field over the last couple of years is the concept of regulatory harmonization. According to experts, there are significant problems in multi-country clinical trials due to the different requirements of the parties involved. A complicating factor is that the definitions of CGT products and their components differ between countries, making it difficult to achieve any consensus on their regulation. Harmonization of CGT requirements and improved inter-country communication are key for the progression of the field throughout Europe.
To address this issue, the FDA’s CBER has developed a Collaboration on Gene Therapies Global Pilot (CoGenT Global). CoGenT Global hopes to provide opportunities for the collaborative review of gene therapy products by international regulators. This includes allowing foreign regulators to join internal FDA meetings, share information, and work together on reviews.
Lack of agreement on surrogate endpoints in clinical trials is another key theme that researchers and regulators hope to resolve. The FDA’s accelerated approval program, which allows for earlier approval of new therapies that address unmet medical needs, is based on surrogate endpoints. These endpoints are markers that can predict clinical benefit but are not actual measurements of clinical benefit as would be expected from standard trials.
Former director of CBER, Peter Marks, has previously noted that accelerated approval using surrogate endpoints is necessary to develop gene therapy products for rare disease populations, however, the FDA often does not agree to surrogate endpoints being accurate predictors of the clinical benefit of a therapy before the trial begins. The FDA’s final guidance on human gene therapy products incorporating human genome editing has now provided some clarification on the use of surrogate endpoints under its accelerated approval program.
For the most current and in-depth information on the FDA's guidelines for cellular and gene therapy products, please visit their webpage.
A major topic of discussion in the cell and gene therapy field over the last couple of years is the concept of regulatory harmonization. According to experts, there are significant problems in multi-country clinical trials due to the different requirements of the parties involved. A complicating factor is that the definitions of CGT products and their components differ between countries, making it difficult to achieve any consensus on their regulation. Harmonization of CGT requirements and improved inter-country communication are key for the progression of the field throughout Europe.
To address this issue, the FDA’s CBER has developed a Collaboration on Gene Therapies Global Pilot (CoGenT Global). CoGenT Global hopes to provide opportunities for the collaborative review of gene therapy products by international regulators. This includes allowing foreign regulators to join internal FDA meetings, share information, and work together on reviews.
Lack of agreement on surrogate endpoints in clinical trials is another key theme that researchers and regulators hope to resolve. The FDA’s accelerated approval program, which allows for earlier approval of new therapies that address unmet medical needs, is based on surrogate endpoints. These endpoints are markers that can predict clinical benefit but are not actual measurements of clinical benefit as would be expected from standard trials.
Former director of CBER, Peter Marks, has previously noted that accelerated approval using surrogate endpoints is necessary to develop gene therapy products for rare disease populations, however, the FDA often does not agree to surrogate endpoints being accurate predictors of the clinical benefit of a therapy before the trial begins. The FDA’s final guidance on human gene therapy products incorporating human genome editing has now provided some clarification on the use of surrogate endpoints under its accelerated approval program.